Daniel Ye, Tong Wu, Ankita Puri, David D. Hebert, Maxime A. Siegler, Michael P. Hendrich, Marcel Swart, Isaac Garcia-Bosch
{"title":"Enhanced Proton-Coupled Electron-Transfer Reactivity by a Mononuclear Nickel(II) Hydroxide Radical Complex","authors":"Daniel Ye, Tong Wu, Ankita Puri, David D. Hebert, Maxime A. Siegler, Michael P. Hendrich, Marcel Swart, Isaac Garcia-Bosch","doi":"10.1021/acs.inorgchem.4c03370","DOIUrl":null,"url":null,"abstract":"The synthesis, characterization, and reactivity of a NiOH core bearing a tridentate redox-active ligand capable of reaching three molecular oxidation states is presented in this paper. The reduced complex [LNiOH]<sup>2–</sup> was characterized by single-crystal X-ray diffraction analysis, depicting a square-planar NiOH core stabilized by intramolecular H-bonding interactions. Cyclic voltammetry measurements indicated that [LNiOH]<sup>2–</sup> can be reversibly oxidized to [LNiOH]<sup>−</sup> and [LNiOH] at very negative reduction potentials (−1.13 and −0.39 V vs ferrocene, respectively). The oxidation of [LNiOH]<sup>2–</sup> to [LNiOH]<sup>−</sup> and [LNiOH] was accomplished using 1 and 2 equiv of ferrocenium, respectively. Spectroscopic and computational characterization suggest that [LNiOH]<sup>2–</sup>, [LNiOH]<sup>−</sup>, and [LNiOH] are all Ni<sup>II</sup> species in which the redox-active ligand adopts different oxidation states (catecholate-like, semiquinone-like, and quinone-like, respectively). The NiOH species were found to promote H-atom abstraction from organic substrates, with [LNiOH]<sup>−</sup> acting as a 1H<sup>+</sup>/1e<sup>–</sup> oxidant and [LNiOH] as a 2H<sup>+</sup>/2e<sup>–</sup> oxidant. Thermochemical analysis indicated that [LNiOH] was capable of abstracting H atoms from stronger O–H bonds than [LNiOH]<sup>−</sup>. However, the greater thermochemical tendency of [LNiOH] reactivity toward H atoms did not align with the kinetics of the PCET reaction, where [LNiOH]<sup>−</sup> reacted with H-atom donors much faster than [LNiOH]. The unique stereoelectronic structure of [LNiOH]<sup>−</sup> (radical character combined with a basic NiOH core) might account for its enhanced PCET reactivity.","PeriodicalId":40,"journal":{"name":"Inorganic Chemistry","volume":"18 1","pages":""},"PeriodicalIF":4.7000,"publicationDate":"2024-12-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.inorgchem.4c03370","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
Abstract
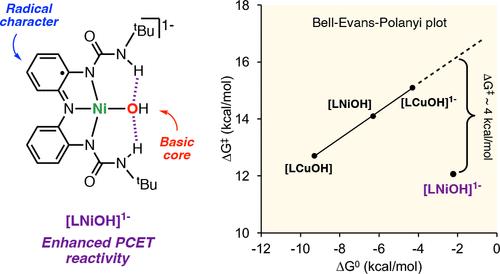
The synthesis, characterization, and reactivity of a NiOH core bearing a tridentate redox-active ligand capable of reaching three molecular oxidation states is presented in this paper. The reduced complex [LNiOH]2– was characterized by single-crystal X-ray diffraction analysis, depicting a square-planar NiOH core stabilized by intramolecular H-bonding interactions. Cyclic voltammetry measurements indicated that [LNiOH]2– can be reversibly oxidized to [LNiOH]− and [LNiOH] at very negative reduction potentials (−1.13 and −0.39 V vs ferrocene, respectively). The oxidation of [LNiOH]2– to [LNiOH]− and [LNiOH] was accomplished using 1 and 2 equiv of ferrocenium, respectively. Spectroscopic and computational characterization suggest that [LNiOH]2–, [LNiOH]−, and [LNiOH] are all NiII species in which the redox-active ligand adopts different oxidation states (catecholate-like, semiquinone-like, and quinone-like, respectively). The NiOH species were found to promote H-atom abstraction from organic substrates, with [LNiOH]− acting as a 1H+/1e– oxidant and [LNiOH] as a 2H+/2e– oxidant. Thermochemical analysis indicated that [LNiOH] was capable of abstracting H atoms from stronger O–H bonds than [LNiOH]−. However, the greater thermochemical tendency of [LNiOH] reactivity toward H atoms did not align with the kinetics of the PCET reaction, where [LNiOH]− reacted with H-atom donors much faster than [LNiOH]. The unique stereoelectronic structure of [LNiOH]− (radical character combined with a basic NiOH core) might account for its enhanced PCET reactivity.
期刊介绍:
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry. Coverage includes experimental and theoretical reports on quantitative studies of structure and thermodynamics, kinetics, mechanisms of inorganic reactions, bioinorganic chemistry, and relevant aspects of organometallic chemistry, solid-state phenomena, and chemical bonding theory. Emphasis is placed on the synthesis, structure, thermodynamics, reactivity, spectroscopy, and bonding properties of significant new and known compounds.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
