Max C. Gallant, Matthew J. McDermott, Bryant Li, Kristin A. Persson
{"title":"A Cellular Automaton Simulation for Predicting Phase Evolution in Solid-State Reactions","authors":"Max C. Gallant, Matthew J. McDermott, Bryant Li, Kristin A. Persson","doi":"10.1021/acs.chemmater.4c02301","DOIUrl":null,"url":null,"abstract":"New computational tools for solid-state synthesis recipe design are needed in order to accelerate the experimental realization of novel functional materials proposed by high-throughput materials discovery workflows. This work contributes a cellular automaton simulation framework for predicting the time-dependent evolution of intermediate and product phases during solid-state reactions as a function of precursor choice and amount, reaction atmosphere, and heating profile. The simulation captures the effects of reactant particle spatial distribution, particle melting, and reaction atmosphere. Reaction rates based on rudimentary kinetics are estimated using density functional theory data from the Materials Project and machine learning estimators for the melting point and the vibrational entropy component of the Gibbs free energy. The resulting simulation framework allows for the prediction of the likely outcome of a reaction recipe before any experiments are performed. We analyze five experimental solid-state recipes for BaTiO<sub>3</sub>, CaZrN<sub>2</sub>, and YMnO<sub>3</sub> found in the literature to illustrate the performance of the model in capturing reaction selectivity and reaction pathways as a function of temperature and precursor choice. This simulation framework offers an easier way to optimize existing recipes, aid in the identification of intermediates, and design effective recipes for yet unrealized inorganic solids <i>in silico</i>.","PeriodicalId":33,"journal":{"name":"Chemistry of Materials","volume":"47 1","pages":""},"PeriodicalIF":7.0000,"publicationDate":"2024-12-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemistry of Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1021/acs.chemmater.4c02301","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
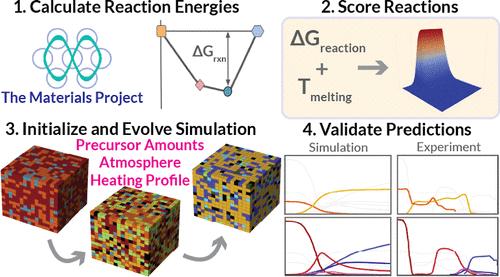
New computational tools for solid-state synthesis recipe design are needed in order to accelerate the experimental realization of novel functional materials proposed by high-throughput materials discovery workflows. This work contributes a cellular automaton simulation framework for predicting the time-dependent evolution of intermediate and product phases during solid-state reactions as a function of precursor choice and amount, reaction atmosphere, and heating profile. The simulation captures the effects of reactant particle spatial distribution, particle melting, and reaction atmosphere. Reaction rates based on rudimentary kinetics are estimated using density functional theory data from the Materials Project and machine learning estimators for the melting point and the vibrational entropy component of the Gibbs free energy. The resulting simulation framework allows for the prediction of the likely outcome of a reaction recipe before any experiments are performed. We analyze five experimental solid-state recipes for BaTiO3, CaZrN2, and YMnO3 found in the literature to illustrate the performance of the model in capturing reaction selectivity and reaction pathways as a function of temperature and precursor choice. This simulation framework offers an easier way to optimize existing recipes, aid in the identification of intermediates, and design effective recipes for yet unrealized inorganic solids in silico.
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
