Sven Larsen-Ledet, Søren Lindemose, Aleksandra Panfilova, Sarah Gersing, Caroline H. Suhr, Aitana Victoria Genzor, Heleen Lanters, Sofie V. Nielsen, Kresten Lindorff-Larsen, Jakob R. Winther, Amelie Stein, Rasmus Hartmann-Petersen
{"title":"Systematic characterization of indel variants using a yeast-based protein folding sensor","authors":"Sven Larsen-Ledet, Søren Lindemose, Aleksandra Panfilova, Sarah Gersing, Caroline H. Suhr, Aitana Victoria Genzor, Heleen Lanters, Sofie V. Nielsen, Kresten Lindorff-Larsen, Jakob R. Winther, Amelie Stein, Rasmus Hartmann-Petersen","doi":"10.1016/j.str.2024.11.017","DOIUrl":null,"url":null,"abstract":"Gene variants resulting in insertions or deletions of amino acid residues (indels) have important consequences for evolution and are often linked to disease, yet, compared to missense variants, the effects of indels are poorly understood and predicted. We developed a sensitive protein folding sensor based on the complementation of uracil auxotrophy in yeast by circular permutated orotate phosphoribosyltransferase (CPOP). The sensor reports on the folding of disease-linked missense variants and <em>de</em>-<em>novo</em>-designed proteins. Applying the folding sensor to a saturated library of single-residue indels in human dihydrofolate reductase (DHFR) revealed that most regions that tolerate indels are confined to internal loops, the termini, and a central α helix. Several indels are temperature sensitive, and folding is rescued upon binding to methotrexate. Rosetta and AlphaFold2 predictions correlate with the observed effects, suggesting that most indels destabilize the native fold and that these computational tools are useful for the classification of indels observed in population sequencing.","PeriodicalId":22168,"journal":{"name":"Structure","volume":"87 1","pages":""},"PeriodicalIF":4.3000,"publicationDate":"2024-12-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structure","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.str.2024.11.017","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
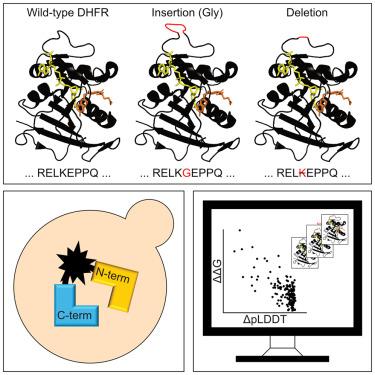
Gene variants resulting in insertions or deletions of amino acid residues (indels) have important consequences for evolution and are often linked to disease, yet, compared to missense variants, the effects of indels are poorly understood and predicted. We developed a sensitive protein folding sensor based on the complementation of uracil auxotrophy in yeast by circular permutated orotate phosphoribosyltransferase (CPOP). The sensor reports on the folding of disease-linked missense variants and de-novo-designed proteins. Applying the folding sensor to a saturated library of single-residue indels in human dihydrofolate reductase (DHFR) revealed that most regions that tolerate indels are confined to internal loops, the termini, and a central α helix. Several indels are temperature sensitive, and folding is rescued upon binding to methotrexate. Rosetta and AlphaFold2 predictions correlate with the observed effects, suggesting that most indels destabilize the native fold and that these computational tools are useful for the classification of indels observed in population sequencing.
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
