Xinbin Ye, Shiwei Hu, Guan Zhang, Yabin Yan, Quanhua Sun, Yuan Hu
{"title":"Uncover Chemical Processes for Silica Surfaces Exposed to Atomic Oxygen Using ReaxFF Reactive Molecular Dynamics","authors":"Xinbin Ye, Shiwei Hu, Guan Zhang, Yabin Yan, Quanhua Sun, Yuan Hu","doi":"10.1021/acs.jpcc.4c05596","DOIUrl":null,"url":null,"abstract":"The considerable level of uncertainty in the measured and calculated catalytic recombination coefficients of atomic oxygen (O) on silica (SiO<sub>2</sub>) surfaces has posed a great challenge to the accurate prediction of heating load and thereby the weight-effective design for atmospheric hypersonic vehicles. This work conducts large-scale (in terms of reaction trajectories) reactive molecular dynamics simulations based on ReaxFF<sub>SiO</sub><sup>GSI</sup>, a ReaxFF potential function tailored for O(gas)-SiO<sub>2</sub>(surface) interactions to understand the chemical processes for the recombination of O for different SiO<sub>2</sub> surface structures. The applicability of the present ReaxFF-based molecular dynamics is validated by the density-functional-theory calculation through O adsorption on the same SiO<sub>2</sub> surface structures under investigation. An automatic data analyzer is developed to capture the reaction pathways and mechanisms from the vast amount of trajectories. It is found that the pathways of adsorption, active site formation, and recombination are sensitive to the surface structures. The overall recombination coefficient and its compositions from different reaction pathways vary considerably for different surface structures. We identify for the first time a reaction mechanism involving multiple active sites, which is more likely to occur than the single-site reactions and thus can potentially increase the recombination probability. These findings highlight the important role of surface structure in catalytic recombination reactions and provide a possible explanation for the huge discrepancy in the recombination coefficients from previous studies.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"23 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-12-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c05596","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
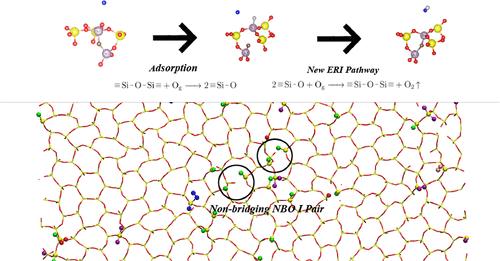
The considerable level of uncertainty in the measured and calculated catalytic recombination coefficients of atomic oxygen (O) on silica (SiO2) surfaces has posed a great challenge to the accurate prediction of heating load and thereby the weight-effective design for atmospheric hypersonic vehicles. This work conducts large-scale (in terms of reaction trajectories) reactive molecular dynamics simulations based on ReaxFFSiOGSI, a ReaxFF potential function tailored for O(gas)-SiO2(surface) interactions to understand the chemical processes for the recombination of O for different SiO2 surface structures. The applicability of the present ReaxFF-based molecular dynamics is validated by the density-functional-theory calculation through O adsorption on the same SiO2 surface structures under investigation. An automatic data analyzer is developed to capture the reaction pathways and mechanisms from the vast amount of trajectories. It is found that the pathways of adsorption, active site formation, and recombination are sensitive to the surface structures. The overall recombination coefficient and its compositions from different reaction pathways vary considerably for different surface structures. We identify for the first time a reaction mechanism involving multiple active sites, which is more likely to occur than the single-site reactions and thus can potentially increase the recombination probability. These findings highlight the important role of surface structure in catalytic recombination reactions and provide a possible explanation for the huge discrepancy in the recombination coefficients from previous studies.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
