{"title":"TSS-Captur: a user-friendly pipeline for characterizing unclassified RNA transcripts.","authors":"Mathias Witte Paz, Thomas Vogel, Kay Nieselt","doi":"10.1093/nargab/lqae168","DOIUrl":null,"url":null,"abstract":"<p><p>RNA-seq and its 5'-enrichment methods for prokaryotes have enabled the precise identification of transcription start sites (TSSs), improving gene expression analysis. Computational methods are applied to these data to identify TSSs and classify them based on proximal annotated genes. While some TSSs cannot be classified at all (orphan TSSs), other TSSs are found on the reverse strand of known genes (antisense TSSs) but are not associated with the direct transcription of any known gene. Here, we introduce TSS-Captur, a novel pipeline, which uses computational approaches to characterize genomic regions starting from experimentally confirmed but unclassified TSSs. By analyzing TSS data, TSS-Captur characterizes unclassified signals, complementing prokaryotic genome annotation tools. TSS-Captur categorizes extracted transcripts as either messenger RNA for genes with coding potential or non-coding RNA (ncRNA) for non-translated genes. Additionally, it predicts the transcription termination site for each putative transcript. For ncRNA genes, the secondary structure is computed. Moreover, all putative promoter regions are analyzed to identify enriched motifs. An interactive report allows seamless data exploration. We validated TSS-Captur with a <i>Campylobacter jejuni</i> dataset and characterized unlabeled ncRNAs in <i>Streptomyces coelicolor</i>. TSS-Captur is available both as a web-application and as a command-line tool.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"6 4","pages":"lqae168"},"PeriodicalIF":2.8000,"publicationDate":"2024-12-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11655288/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqae168","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
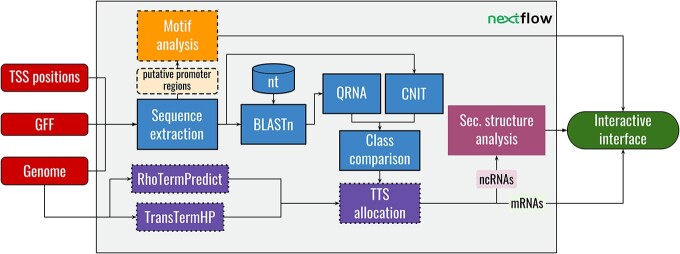
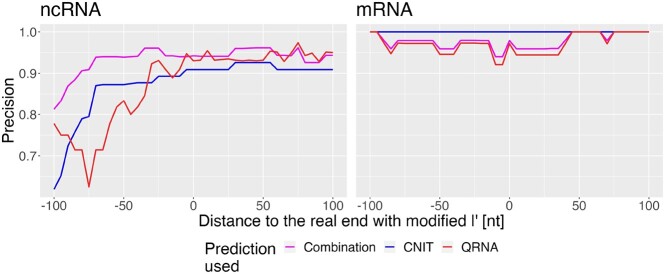
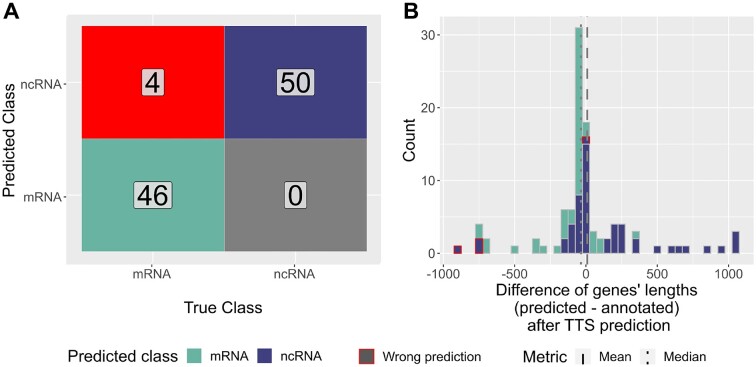
RNA-seq and its 5'-enrichment methods for prokaryotes have enabled the precise identification of transcription start sites (TSSs), improving gene expression analysis. Computational methods are applied to these data to identify TSSs and classify them based on proximal annotated genes. While some TSSs cannot be classified at all (orphan TSSs), other TSSs are found on the reverse strand of known genes (antisense TSSs) but are not associated with the direct transcription of any known gene. Here, we introduce TSS-Captur, a novel pipeline, which uses computational approaches to characterize genomic regions starting from experimentally confirmed but unclassified TSSs. By analyzing TSS data, TSS-Captur characterizes unclassified signals, complementing prokaryotic genome annotation tools. TSS-Captur categorizes extracted transcripts as either messenger RNA for genes with coding potential or non-coding RNA (ncRNA) for non-translated genes. Additionally, it predicts the transcription termination site for each putative transcript. For ncRNA genes, the secondary structure is computed. Moreover, all putative promoter regions are analyzed to identify enriched motifs. An interactive report allows seamless data exploration. We validated TSS-Captur with a Campylobacter jejuni dataset and characterized unlabeled ncRNAs in Streptomyces coelicolor. TSS-Captur is available both as a web-application and as a command-line tool.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
