Sevinç Odabaşı Güneş, Havva Nur Peltek Kendirci, Edip Ünal, Ayşe Derya Buluş, İsmail Dündar, Zeynep Şıklar
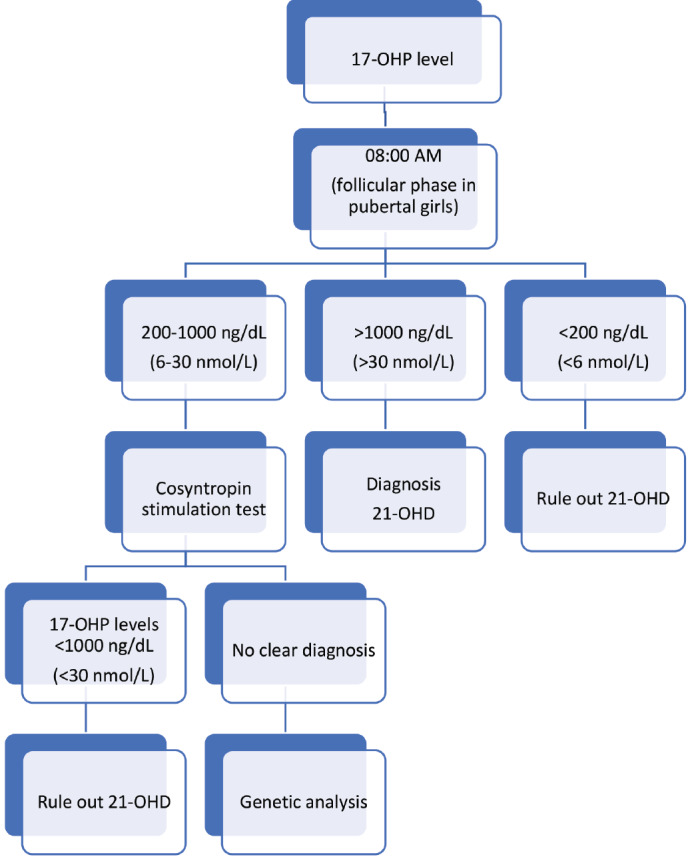
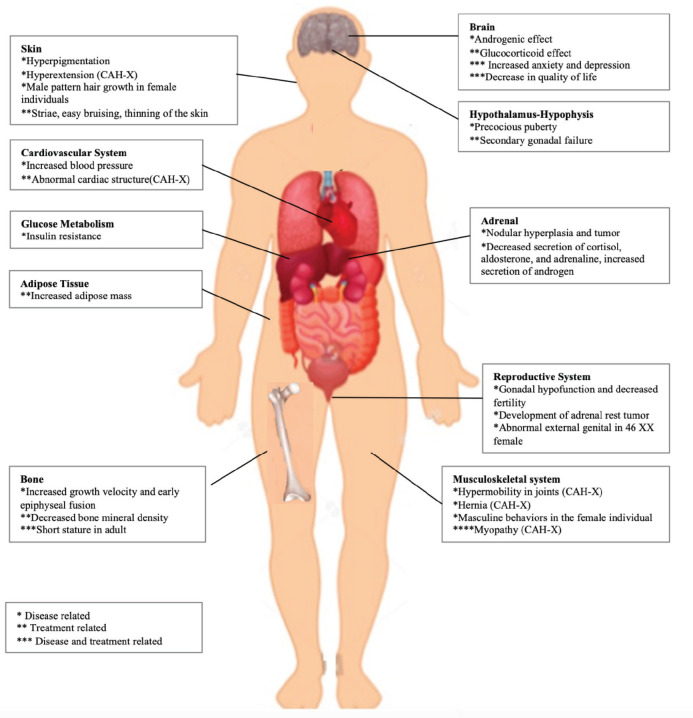
{"title":"Clinical, Biochemical and Molecular Characteristics of Congenital Adrenal Hyperplasia Due to 21-hydroxylase Deficiency","authors":"Sevinç Odabaşı Güneş, Havva Nur Peltek Kendirci, Edip Ünal, Ayşe Derya Buluş, İsmail Dündar, Zeynep Şıklar","doi":"10.4274/jcrpe.galenos.2024.2024-6-6-S","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital adrenal hyperplasia (CAH) is an autosomal recessive disease caused by the deficiency of one of the enzymes involved in cortisol synthesis. Between 90% and 99% of cases of CAH are caused by 21-hydroxylase deficiency (21-OHD) caused by mutations in <i>CYP21A2</i>. Although 21-OHD has been historically divided into classical and non-classical forms, it is now thought to show a continuous phenotype. In the classical form, the external genitalia in females becomes virilized to varying degrees. If the disease is not recognized, salt wasting crises in the classical form may threaten life in neonates. Children experience accelerated somatic growth, increased bone age, and premature pubic hair in the simple virilizing form of classical 21-OHD. Female adolescents may present with severe acne, hirsutism, androgenic alopecia, menstrual irregularity or primary amenorrhea in the non-classical form. Diagnosis of CAH is made by clinical, biochemical and molecular genetic evaluation. In cases of 21-OHD, the diagnosis is based on the 17-hydroxyprogesterone (17-OHP) level being above 1000 ng/dL, measured early in the morning. In cases with borderline 17-OHP levels (200-1000 ng/dL), it is recommended to perform an adrenocorticotropic hormone (ACTH) stimulation test. Genotyping in cases with CAH should be performed if the adrenocortical profile is suspicious or if the ACTH stimulation test cannot be performed completely. After diagnosis, determining the carrier status of the parents and determining which parent the mutation was passed on from will help in interpreting the genetic results and determining the risk of recurrence in subsequent pregnancies.</p>","PeriodicalId":48805,"journal":{"name":"Journal of Clinical Research in Pediatric Endocrinology","volume":" ","pages":"3-11"},"PeriodicalIF":1.5000,"publicationDate":"2025-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11730093/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Research in Pediatric Endocrinology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.4274/jcrpe.galenos.2024.2024-6-6-S","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/23 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disease caused by the deficiency of one of the enzymes involved in cortisol synthesis. Between 90% and 99% of cases of CAH are caused by 21-hydroxylase deficiency (21-OHD) caused by mutations in CYP21A2. Although 21-OHD has been historically divided into classical and non-classical forms, it is now thought to show a continuous phenotype. In the classical form, the external genitalia in females becomes virilized to varying degrees. If the disease is not recognized, salt wasting crises in the classical form may threaten life in neonates. Children experience accelerated somatic growth, increased bone age, and premature pubic hair in the simple virilizing form of classical 21-OHD. Female adolescents may present with severe acne, hirsutism, androgenic alopecia, menstrual irregularity or primary amenorrhea in the non-classical form. Diagnosis of CAH is made by clinical, biochemical and molecular genetic evaluation. In cases of 21-OHD, the diagnosis is based on the 17-hydroxyprogesterone (17-OHP) level being above 1000 ng/dL, measured early in the morning. In cases with borderline 17-OHP levels (200-1000 ng/dL), it is recommended to perform an adrenocorticotropic hormone (ACTH) stimulation test. Genotyping in cases with CAH should be performed if the adrenocortical profile is suspicious or if the ACTH stimulation test cannot be performed completely. After diagnosis, determining the carrier status of the parents and determining which parent the mutation was passed on from will help in interpreting the genetic results and determining the risk of recurrence in subsequent pregnancies.
期刊介绍:
The Journal of Clinical Research in Pediatric Endocrinology (JCRPE) publishes original research articles, reviews, short communications, letters, case reports and other special features related to the field of pediatric endocrinology. JCRPE is published in English by the Turkish Pediatric Endocrinology and Diabetes Society quarterly (March, June, September, December). The target audience is physicians, researchers and other healthcare professionals in all areas of pediatric endocrinology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
