Will C. Hartley, Kevin Kasten, Mark D. Greenhalgh, Taisiia Feoktistova, Henry R. Wise, Jacqueline M. Laddusaw, Aileen B. Frost, Sean Ng, Alexandra M. Z. Slawin, Bela E. Bode, Paul Ha-Yeon Cheong, Andrew D. Smith
{"title":"In-Cage Recombination Facilitates the Enantioselective Organocatalytic [1,2]-Rearrangement of Allylic Ammonium Ylides","authors":"Will C. Hartley, Kevin Kasten, Mark D. Greenhalgh, Taisiia Feoktistova, Henry R. Wise, Jacqueline M. Laddusaw, Aileen B. Frost, Sean Ng, Alexandra M. Z. Slawin, Bela E. Bode, Paul Ha-Yeon Cheong, Andrew D. Smith","doi":"10.1021/jacs.4c14516","DOIUrl":null,"url":null,"abstract":"The [1,2]-rearrangement of allylic ammonium ylides is traditionally observed as a competitive minor pathway alongside the thermally allowed [2,3]-sigmatropic rearrangement. Concerted [1,2]-rearrangements are formally forbidden, with these processes believed to proceed through homolytic C–N bond fission of the ylide, followed by radical–radical recombination. The challenges associated with developing a catalytic enantioselective [1,2]-rearrangement of allylic ammonium ylides therefore lie in biasing the reaction pathway to favor the [1,2]-reaction product, alongside controlling a stereoselective radical–radical recombination event. Herein, a Lewis basic chiral isothiourea facilitates catalytic [1,2]-rearrangement of prochiral aryl ester ammonium salts to generate unnatural α-amino acid derivatives with up to complete selectivity over the [2,3]-rearrangement and with good to excellent enantiocontrol. Key factors in favoring the [1,2]-rearrangement include exploitation of disubstituted terminal allylic substituents, cyclic N-substituted ammonium salts, and elevated reaction temperatures. Mechanistic studies involving <sup>13</sup>C-labeling and crossover reactions, combined with radical trapping experiments and observed changes in product enantioselectivity, are consistent with a radical solvent cage effect, with maximum product enantioselectivity observed through promotion of “in-cage” radical–radical recombination. Computational analysis indicates that the distribution between [1,2]- and [2,3]-rearrangement products arises predominantly from C–N bond homolysis of an intermediate ammonium ylide, followed by recombination of the α-amino radical at either the primary or tertiary site of an intermediate allylic radical. Electrostatic interactions involving the bromide counterion control the facial selectivity of the [1,2]- and [2,3]-rearrangements, while the sterically hindered tertiary position of the allylic substituent disfavors the formation of the [2,3]-product. These results will impact further investigations and understanding of enantioselective radical–radical reactions.","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"28 1","pages":""},"PeriodicalIF":15.6000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/jacs.4c14516","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
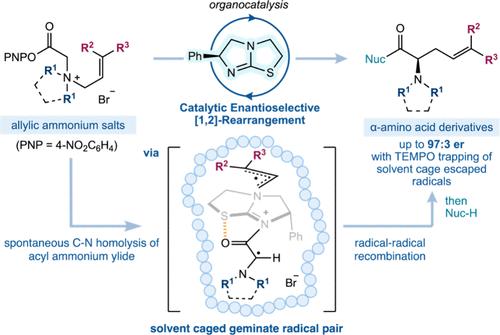
The [1,2]-rearrangement of allylic ammonium ylides is traditionally observed as a competitive minor pathway alongside the thermally allowed [2,3]-sigmatropic rearrangement. Concerted [1,2]-rearrangements are formally forbidden, with these processes believed to proceed through homolytic C–N bond fission of the ylide, followed by radical–radical recombination. The challenges associated with developing a catalytic enantioselective [1,2]-rearrangement of allylic ammonium ylides therefore lie in biasing the reaction pathway to favor the [1,2]-reaction product, alongside controlling a stereoselective radical–radical recombination event. Herein, a Lewis basic chiral isothiourea facilitates catalytic [1,2]-rearrangement of prochiral aryl ester ammonium salts to generate unnatural α-amino acid derivatives with up to complete selectivity over the [2,3]-rearrangement and with good to excellent enantiocontrol. Key factors in favoring the [1,2]-rearrangement include exploitation of disubstituted terminal allylic substituents, cyclic N-substituted ammonium salts, and elevated reaction temperatures. Mechanistic studies involving 13C-labeling and crossover reactions, combined with radical trapping experiments and observed changes in product enantioselectivity, are consistent with a radical solvent cage effect, with maximum product enantioselectivity observed through promotion of “in-cage” radical–radical recombination. Computational analysis indicates that the distribution between [1,2]- and [2,3]-rearrangement products arises predominantly from C–N bond homolysis of an intermediate ammonium ylide, followed by recombination of the α-amino radical at either the primary or tertiary site of an intermediate allylic radical. Electrostatic interactions involving the bromide counterion control the facial selectivity of the [1,2]- and [2,3]-rearrangements, while the sterically hindered tertiary position of the allylic substituent disfavors the formation of the [2,3]-product. These results will impact further investigations and understanding of enantioselective radical–radical reactions.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
