{"title":"Multistate Transition Metal Carbonyl Bonding Beyond Minimum Energy Pathway: Nonlocality of Spin–Orbit Interaction","authors":"Daisuke Yoshida, Kaito Takahashi","doi":"10.1021/acs.inorgchem.4c04568","DOIUrl":null,"url":null,"abstract":"Transition metal carbonyl and transition metal dinitrogen are fundamental chemical complexes in many important biological and catalytic processes. Interestingly, binding between a transition metal (TM) atom and carbonyl or dinitrogen results in spin state change. However, no study has evaluated the spin–orbit (SO) effect along the association pathway of any TM–CO or TM–N<sub>2</sub> bond. Using multireference calculations with SO interaction, we calculated the association potential energy curve for 11 electronic states for the spin crossover reactions: Ni + CO → NiCO and Ni + N<sub>2</sub> → NiN<sub>2</sub>. Through this multistate calculation, we found that the commonly used minimum energy pathway (MEP) gives reasonable energies for the asymptotes but has an incorrect physical picture in the intermediate bond length. MEP assumes strong SO at the energy crossing point that allows for direct spin crossover from the triplet to singlet state, but multireference calculations showed that SO interactions strengthen at bond length regions, 2.3–2.5 Å before the crossing point. Furthermore, this results in a spin barrier of 0.15 eV along the Ni adsorbate association pathway. These calculations provide a new understanding of the overlooked yet important effect of the spin barrier on the association process, which can change the association rate by several orders of magnitude.","PeriodicalId":40,"journal":{"name":"Inorganic Chemistry","volume":"69 1","pages":""},"PeriodicalIF":4.7000,"publicationDate":"2024-12-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.inorgchem.4c04568","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
Abstract
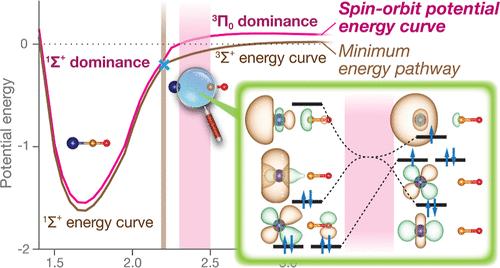
Transition metal carbonyl and transition metal dinitrogen are fundamental chemical complexes in many important biological and catalytic processes. Interestingly, binding between a transition metal (TM) atom and carbonyl or dinitrogen results in spin state change. However, no study has evaluated the spin–orbit (SO) effect along the association pathway of any TM–CO or TM–N2 bond. Using multireference calculations with SO interaction, we calculated the association potential energy curve for 11 electronic states for the spin crossover reactions: Ni + CO → NiCO and Ni + N2 → NiN2. Through this multistate calculation, we found that the commonly used minimum energy pathway (MEP) gives reasonable energies for the asymptotes but has an incorrect physical picture in the intermediate bond length. MEP assumes strong SO at the energy crossing point that allows for direct spin crossover from the triplet to singlet state, but multireference calculations showed that SO interactions strengthen at bond length regions, 2.3–2.5 Å before the crossing point. Furthermore, this results in a spin barrier of 0.15 eV along the Ni adsorbate association pathway. These calculations provide a new understanding of the overlooked yet important effect of the spin barrier on the association process, which can change the association rate by several orders of magnitude.
期刊介绍:
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry. Coverage includes experimental and theoretical reports on quantitative studies of structure and thermodynamics, kinetics, mechanisms of inorganic reactions, bioinorganic chemistry, and relevant aspects of organometallic chemistry, solid-state phenomena, and chemical bonding theory. Emphasis is placed on the synthesis, structure, thermodynamics, reactivity, spectroscopy, and bonding properties of significant new and known compounds.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
