Mahmoud A.S. Sakr , Hazem Abdelsalam , Nahed H. Teleb , Mohamed A. Saad , Omar H. Abd-Elkader , Yushen Liu , Qinfang Zhang
{"title":"Exploring the drug delivery capabilities of Nb2C MXene functionalized with oxygen and fluorine: A DFT study","authors":"Mahmoud A.S. Sakr , Hazem Abdelsalam , Nahed H. Teleb , Mohamed A. Saad , Omar H. Abd-Elkader , Yushen Liu , Qinfang Zhang","doi":"10.1016/j.jmgm.2024.108937","DOIUrl":null,"url":null,"abstract":"<div><div>MXenes quantum dots (QDs), including Nb<sub>2</sub>C, Nb<sub>2</sub>CO<sub>2</sub>, and Nb<sub>2</sub>CF<sub>2</sub>, are emerging materials with exceptional structural, electronic, and optical properties, making them highly suitable for biomedical applications. This study investigates the structural optimization, stability, electronic properties, and drug-loading potential of these QDs using fluorouracil (Flu) as a model drug. Structural analyses show that the functionalization of Nb<sub>2</sub>C with O and F atoms enhances stability, with binding energies (BEs) of 7.335, 8.154, and 6.704 eV for Nb<sub>2</sub>C, Nb<sub>2</sub>CO<sub>2</sub>, and Nb<sub>2</sub>CF<sub>2</sub>, respectively. The drug-loading study reveals that Nb<sub>2</sub>C exhibits the highest adsorption energy of −6.775 eV at the surface site (2.053 Å), while Nb<sub>2</sub>CO<sub>2</sub> and Nb<sub>2</sub>CF<sub>2</sub> demonstrate weaker interactions with adsorption energies of −2.163 eV and −0.933 eV, respectively. Non-covalent interaction (NCI) and natural bond orbital (NBO) analyses show significant changes in electron density distribution upon drug interaction, with the natural charge on the O7 atom in Flu shifting slightly upon interaction. Optical property investigations indicate a blue shift in the absorption spectra for Nb<sub>2</sub>CO<sub>2</sub> (<em>λ</em><sub>max</sub> = 764.76 nm) and Nb<sub>2</sub>CF<sub>2</sub> (<em>λ</em><sub>max</sub> = 1108.71 nm), compared to Nb<sub>2</sub>C (<em>λ</em><sub>max</sub> = 2612.00 nm), confirming the tunability of these materials for therapeutic applications. By addressing key challenges in drug delivery, such as stability, controlled release, and interaction strength, this study establishes Nb<sub>2</sub>CO<sub>2</sub> and Nb<sub>2</sub>CF<sub>2</sub> as promising nanocarriers, with the potential to improve drug efficacy and minimize side effects in targeted cancer therapies.</div></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"136 ","pages":"Article 108937"},"PeriodicalIF":3.0000,"publicationDate":"2025-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324002377","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
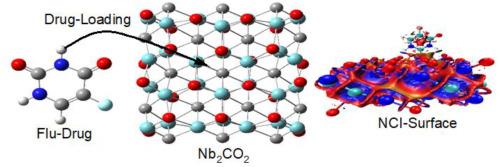
MXenes quantum dots (QDs), including Nb2C, Nb2CO2, and Nb2CF2, are emerging materials with exceptional structural, electronic, and optical properties, making them highly suitable for biomedical applications. This study investigates the structural optimization, stability, electronic properties, and drug-loading potential of these QDs using fluorouracil (Flu) as a model drug. Structural analyses show that the functionalization of Nb2C with O and F atoms enhances stability, with binding energies (BEs) of 7.335, 8.154, and 6.704 eV for Nb2C, Nb2CO2, and Nb2CF2, respectively. The drug-loading study reveals that Nb2C exhibits the highest adsorption energy of −6.775 eV at the surface site (2.053 Å), while Nb2CO2 and Nb2CF2 demonstrate weaker interactions with adsorption energies of −2.163 eV and −0.933 eV, respectively. Non-covalent interaction (NCI) and natural bond orbital (NBO) analyses show significant changes in electron density distribution upon drug interaction, with the natural charge on the O7 atom in Flu shifting slightly upon interaction. Optical property investigations indicate a blue shift in the absorption spectra for Nb2CO2 (λmax = 764.76 nm) and Nb2CF2 (λmax = 1108.71 nm), compared to Nb2C (λmax = 2612.00 nm), confirming the tunability of these materials for therapeutic applications. By addressing key challenges in drug delivery, such as stability, controlled release, and interaction strength, this study establishes Nb2CO2 and Nb2CF2 as promising nanocarriers, with the potential to improve drug efficacy and minimize side effects in targeted cancer therapies.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
