Moriya Iwaizumi, Terumi Taniguchi, Risa Kojima, Harumo Osawa, Kyota Tatsuta, Mayu Sakata, Satoshi Osawa, Kiyotaka Kurachi, Ken Sugimoto
{"title":"Two independent families with de novo whole APC gene deletion and intellectual disability: a case report.","authors":"Moriya Iwaizumi, Terumi Taniguchi, Risa Kojima, Harumo Osawa, Kyota Tatsuta, Mayu Sakata, Satoshi Osawa, Kiyotaka Kurachi, Ken Sugimoto","doi":"10.1186/s13053-024-00297-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Familial adenomatous polyposis (FAP) is an autosomal dominant colorectal tumour syndrome characterised by the formation of multiple adenomatous polyps throughout the colon. It is important to understand the extracolonic phenotype that characterizes FAP. Most previous case reports of patients with both FAP and intellectual disability (ID) have described deletions in all or part of chromosome 5q, including the APC locus. However, it remains unclear whether the ID phenotype in patients with FAP is due to APC disruption or another genetic defect in the deleted 5q region.</p><p><strong>Case presentation: </strong>Patient of family 1 is a 32-year-old woman presented with > 500 colorectal adenomatous polyps, gastric fundic gland polyposis, several duodenal adenomas, and mild intellectual disability (ID). She had no known family history of the FAP phenotype or ID. By copy number trio analysis, a 15.4 Mb interstitial heterozygous de novo deletion including APC region was observed in 5q21.2. q22.3. The patient in family 2 was a 29-year-old man with approximately 50 colorectal adenomatous polyps, fundic gland polyposis in the stomach, non-ampullary adenomas in the duodenum, and mild ID. He had no family history of the FAP phenotype or ID. Using copy number trio analysis, a de novo 9.8 Mb heterozygous deletion was identified on 5q22.1. q23.1 which includes the APC region.</p><p><strong>Conclusions: </strong>Based on previous reports and the present study, we narrowed down the 5p deletion region associated with ID in FAP. Further investigation is required to understand ID due to 5q stromal deletion.</p>","PeriodicalId":55058,"journal":{"name":"Hereditary Cancer in Clinical Practice","volume":"23 1","pages":"1"},"PeriodicalIF":2.4000,"publicationDate":"2025-01-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11708175/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditary Cancer in Clinical Practice","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13053-024-00297-1","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Familial adenomatous polyposis (FAP) is an autosomal dominant colorectal tumour syndrome characterised by the formation of multiple adenomatous polyps throughout the colon. It is important to understand the extracolonic phenotype that characterizes FAP. Most previous case reports of patients with both FAP and intellectual disability (ID) have described deletions in all or part of chromosome 5q, including the APC locus. However, it remains unclear whether the ID phenotype in patients with FAP is due to APC disruption or another genetic defect in the deleted 5q region.
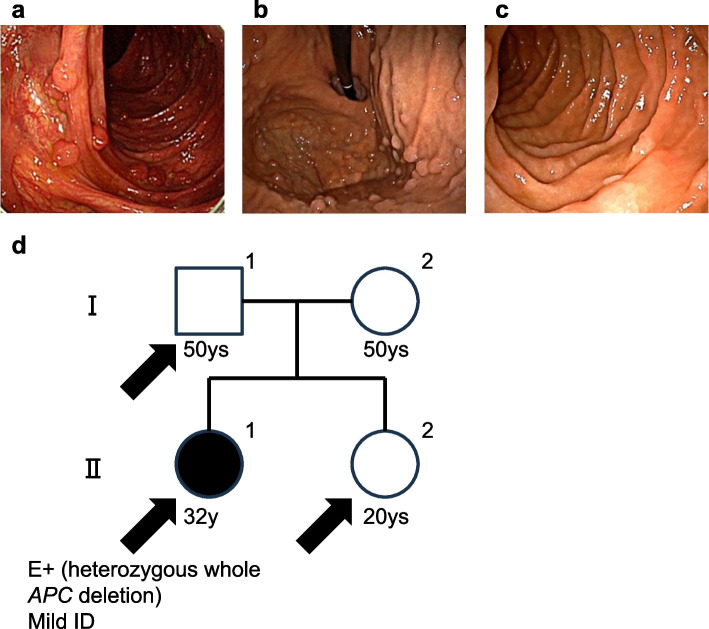
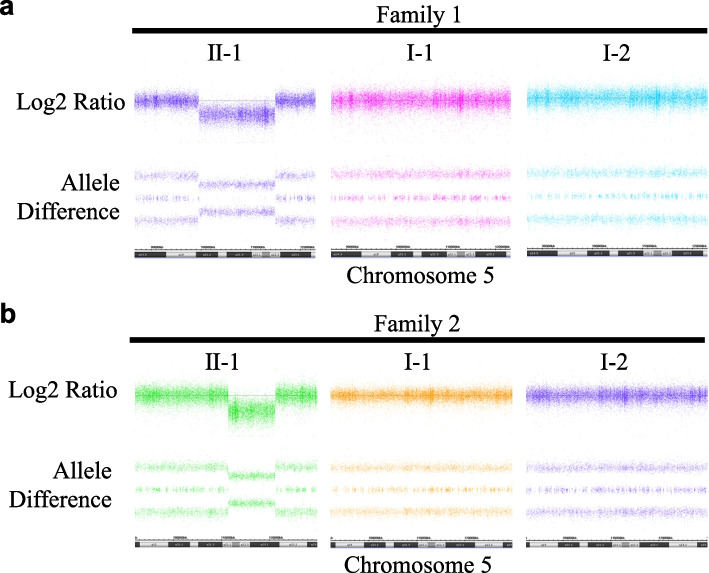
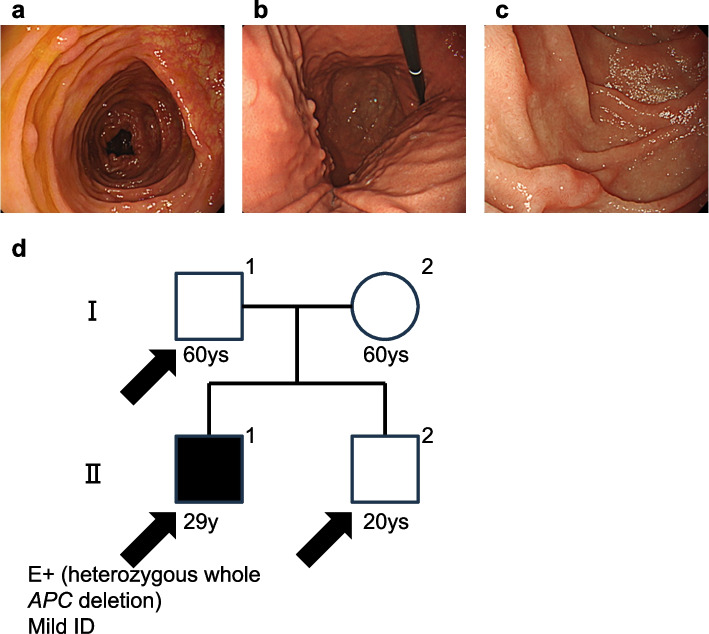
Case presentation: Patient of family 1 is a 32-year-old woman presented with > 500 colorectal adenomatous polyps, gastric fundic gland polyposis, several duodenal adenomas, and mild intellectual disability (ID). She had no known family history of the FAP phenotype or ID. By copy number trio analysis, a 15.4 Mb interstitial heterozygous de novo deletion including APC region was observed in 5q21.2. q22.3. The patient in family 2 was a 29-year-old man with approximately 50 colorectal adenomatous polyps, fundic gland polyposis in the stomach, non-ampullary adenomas in the duodenum, and mild ID. He had no family history of the FAP phenotype or ID. Using copy number trio analysis, a de novo 9.8 Mb heterozygous deletion was identified on 5q22.1. q23.1 which includes the APC region.
Conclusions: Based on previous reports and the present study, we narrowed down the 5p deletion region associated with ID in FAP. Further investigation is required to understand ID due to 5q stromal deletion.
期刊介绍:
Hereditary Cancer in Clinical Practice is an open access journal that publishes articles of interest for the cancer genetics community and serves as a discussion forum for the development appropriate healthcare strategies.
Cancer genetics encompasses a wide variety of disciplines and knowledge in the field is rapidly growing, especially as the amount of information linking genetic differences to inherited cancer predispositions continues expanding. With the increased knowledge of genetic variability and how this relates to cancer risk there is a growing demand not only to disseminate this information into clinical practice but also to enable competent debate concerning how such information is managed and what it implies for patient care.
Topics covered by the journal include but are not limited to:
Original research articles on any aspect of inherited predispositions to cancer.
Reviews of inherited cancer predispositions.
Application of molecular and cytogenetic analysis to clinical decision making.
Clinical aspects of the management of hereditary cancers.
Genetic counselling issues associated with cancer genetics.
The role of registries in improving health care of patients with an inherited predisposition to cancer.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
