Katrin Õunap, Tiia Reimand, Eve Õiglane-Shlik, Sanna Puusepp, Laura Mihkla, Sander Pajusalu, Marco Savarese, Bjarne Udd
{"title":"<i>TTN</i>-Related Muscular Dystrophies, LGMD, and TMD, in an Estonian Family Caused by the Finnish Founder Variant.","authors":"Katrin Õunap, Tiia Reimand, Eve Õiglane-Shlik, Sanna Puusepp, Laura Mihkla, Sander Pajusalu, Marco Savarese, Bjarne Udd","doi":"10.1212/NXG.0000000000200199","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Tibial muscular dystrophy (TMD) is an autosomal dominant, slowly progressive late-onset distal myopathy. TMD was first described in 1991 by Udd et al. in Finnish patients, who were later found to harbor a heterozygous unique 11-bp insertion/deletion in the last exon of the <i>TTN</i> gene-the Finnish founder variant (FINmaj). In homozygous state or compound heterozygosity with a truncating variant, the FINmaj causes early-onset recessive titin-related limb-girdle muscular dystrophy type 10 (LGMD R10). So far, the FINmaj variant has not been detected outside the Finnish population.</p><p><strong>Methods: </strong>We describe an Estonian family presenting both early-onset LGMD R10 and late-onset TMD. The index patient underwent trio exome sequencing (ES), muscle biopsy, and RNA sequencing. The detected variants were validated by Sanger sequencing. Muscle MRI was performed in all affected individuals.</p><p><strong>Results: </strong>Trio ES revealed 2 heterozygous variants in the <i>TTN</i> gene: (NM_001267550.2):c.107780_107790delinsTGAAAGAAAAA, p.(Glu35927_Trp35930delinsValLysGluLys) (FINmaj variant, paternally inherited) and (NM_001267550.2):c.64672+2dup (maternally inherited) in trans in the proband. Familial segregation analysis revealed the same biallelic variants in the younger affected sister and heterozygous FINmaj in the father. We characterized the effect of the splice variant by RNA sequencing, proving that it causes an intronic retention resulting in a premature stop codon. Muscle histology of the proband showed myopathic changes. Muscle MRI of both individuals with LGMD R10 showed early degenerative changes in tibialis anterior and in hypotrophy of distal hamstrings. Muscle MRI of the father with TMD, at the age of 38 years, showed early minimal fatty degeneration in the peroneus longus and right tibialis anterior muscles.</p><p><strong>Discussion: </strong>For the first time, we have detected the FINmaj variant in the Estonian population. We report an Estonian family without any known Finnish ancestry for many generations, with 2 siblings harboring FINmaj in a compound with a splice site variant and their father with heterozygous FINmaj. It is currently not known whether the FINmaj is originally Estonian or Finnish ancestry. Further population studies in Estonia to establish the frequency of FINmaj in the population are ongoing and will solve the quest.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"10 6","pages":"e200199"},"PeriodicalIF":3.7000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11727988/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200199","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objectives: Tibial muscular dystrophy (TMD) is an autosomal dominant, slowly progressive late-onset distal myopathy. TMD was first described in 1991 by Udd et al. in Finnish patients, who were later found to harbor a heterozygous unique 11-bp insertion/deletion in the last exon of the TTN gene-the Finnish founder variant (FINmaj). In homozygous state or compound heterozygosity with a truncating variant, the FINmaj causes early-onset recessive titin-related limb-girdle muscular dystrophy type 10 (LGMD R10). So far, the FINmaj variant has not been detected outside the Finnish population.
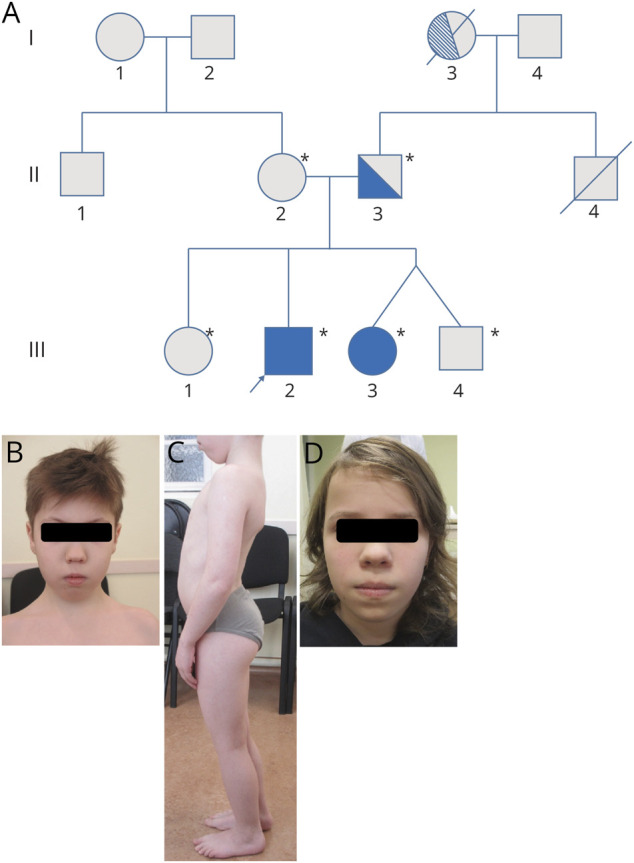
Methods: We describe an Estonian family presenting both early-onset LGMD R10 and late-onset TMD. The index patient underwent trio exome sequencing (ES), muscle biopsy, and RNA sequencing. The detected variants were validated by Sanger sequencing. Muscle MRI was performed in all affected individuals.
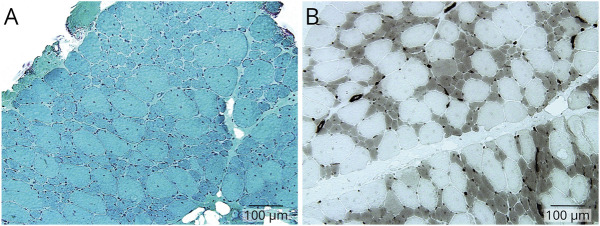
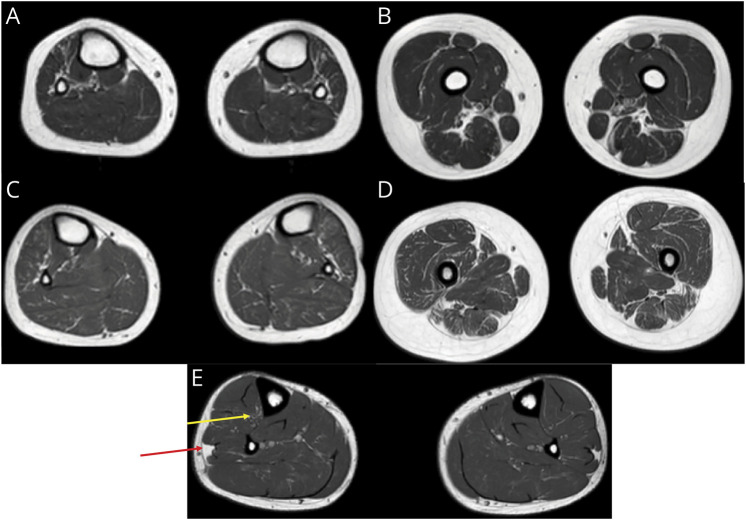
Results: Trio ES revealed 2 heterozygous variants in the TTN gene: (NM_001267550.2):c.107780_107790delinsTGAAAGAAAAA, p.(Glu35927_Trp35930delinsValLysGluLys) (FINmaj variant, paternally inherited) and (NM_001267550.2):c.64672+2dup (maternally inherited) in trans in the proband. Familial segregation analysis revealed the same biallelic variants in the younger affected sister and heterozygous FINmaj in the father. We characterized the effect of the splice variant by RNA sequencing, proving that it causes an intronic retention resulting in a premature stop codon. Muscle histology of the proband showed myopathic changes. Muscle MRI of both individuals with LGMD R10 showed early degenerative changes in tibialis anterior and in hypotrophy of distal hamstrings. Muscle MRI of the father with TMD, at the age of 38 years, showed early minimal fatty degeneration in the peroneus longus and right tibialis anterior muscles.
Discussion: For the first time, we have detected the FINmaj variant in the Estonian population. We report an Estonian family without any known Finnish ancestry for many generations, with 2 siblings harboring FINmaj in a compound with a splice site variant and their father with heterozygous FINmaj. It is currently not known whether the FINmaj is originally Estonian or Finnish ancestry. Further population studies in Estonia to establish the frequency of FINmaj in the population are ongoing and will solve the quest.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
