Bingqing Luo, Xia Wu, Jing Zhu, Shu Chen, Shifeng Lou, Xiaoyan Tan
{"title":"Compound heterozygous mutations (p.L68R∗37 and p.T241N) lead to abnormal protein levels and structures in hereditary FVII deficiency.","authors":"Bingqing Luo, Xia Wu, Jing Zhu, Shu Chen, Shifeng Lou, Xiaoyan Tan","doi":"10.1097/MBC.0000000000001340","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Congenital factor VII (FVII) deficiency is a genetic disorder characterized by decreased FVII activity, which sometimes leads to fatal bleeding. Numerous variants have been found in FVII deficiency, but mutations vary among patients. Each mutation deserves further exploration for each patient at risk of bleeding. We previously reported a Chinese patient with p.L68R∗37 and p.T241N compound heterozygous mutations. In this study, we further investigated the impact of these two mutations on the FVII expression through in vitro expression experiments.</p><p><strong>Methods: </strong>Mutations were introduced into the FVII coding region using site-directed mutagenesis, and recombinant FVII was combined with two different plasmids, and then quantitative PCR and western blot analyses were performed subsequently.</p><p><strong>Results: </strong>The p.L68R∗37 mutation had no effect on mRNA levels but caused a significant decrease in protein levels. In the p.T241N mutant vector, mRNA levels did not show a noticeable decrease, but protein levels exhibited a slight decrease. Structural analysis revealed that the p.T241N mutation resulted in an altered secondary structure and protein instability, indicating impaired functional properties.</p><p><strong>Conclusion: </strong>Our study demonstrated that the p.L68R∗37 and p.T241N mutations impacted the protein levels and function of FVII, ultimately leading to a severe reduction in FVII activity. This study may contribute to further understanding of the molecular pathogenesis of FVII deficiency and offer insights for genetic counseling.</p>","PeriodicalId":8992,"journal":{"name":"Blood Coagulation & Fibrinolysis","volume":" ","pages":"44-50"},"PeriodicalIF":1.1000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11789605/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Blood Coagulation & Fibrinolysis","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1097/MBC.0000000000001340","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/6 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
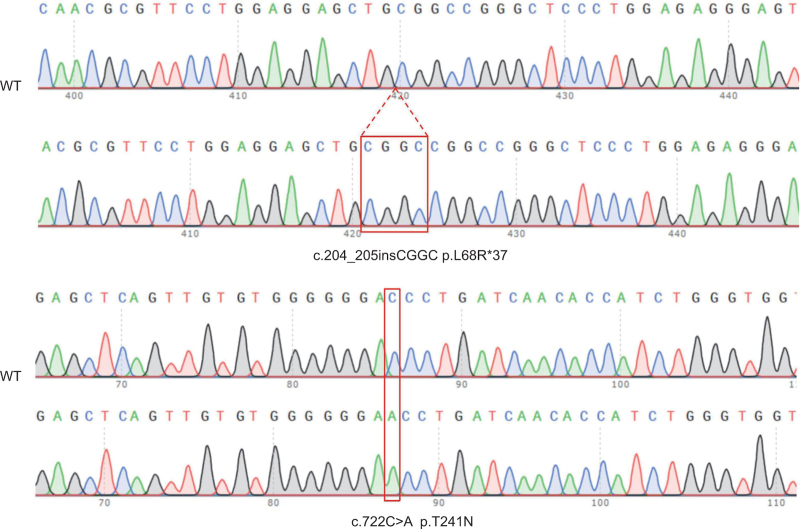
Background: Congenital factor VII (FVII) deficiency is a genetic disorder characterized by decreased FVII activity, which sometimes leads to fatal bleeding. Numerous variants have been found in FVII deficiency, but mutations vary among patients. Each mutation deserves further exploration for each patient at risk of bleeding. We previously reported a Chinese patient with p.L68R∗37 and p.T241N compound heterozygous mutations. In this study, we further investigated the impact of these two mutations on the FVII expression through in vitro expression experiments.
Methods: Mutations were introduced into the FVII coding region using site-directed mutagenesis, and recombinant FVII was combined with two different plasmids, and then quantitative PCR and western blot analyses were performed subsequently.
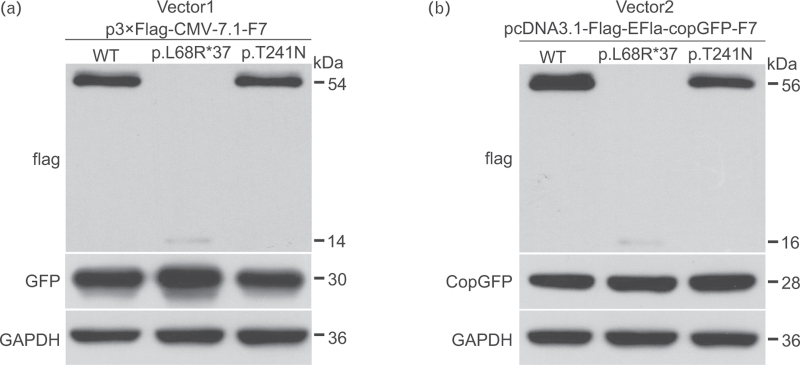
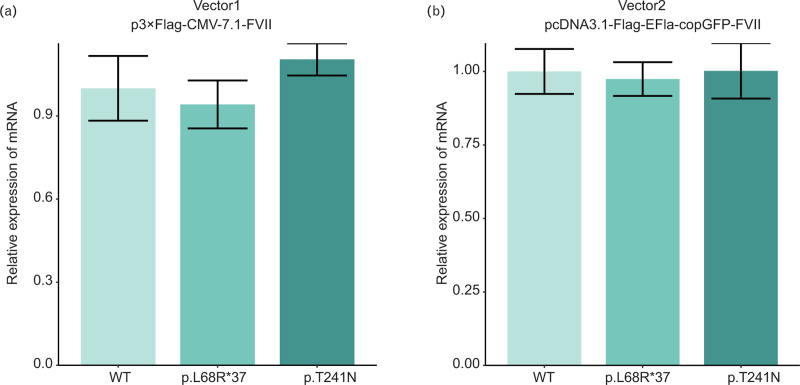
Results: The p.L68R∗37 mutation had no effect on mRNA levels but caused a significant decrease in protein levels. In the p.T241N mutant vector, mRNA levels did not show a noticeable decrease, but protein levels exhibited a slight decrease. Structural analysis revealed that the p.T241N mutation resulted in an altered secondary structure and protein instability, indicating impaired functional properties.
Conclusion: Our study demonstrated that the p.L68R∗37 and p.T241N mutations impacted the protein levels and function of FVII, ultimately leading to a severe reduction in FVII activity. This study may contribute to further understanding of the molecular pathogenesis of FVII deficiency and offer insights for genetic counseling.
期刊介绍:
Blood Coagulation & Fibrinolysis is an international fully refereed journal that features review and original research articles on all clinical, laboratory and experimental aspects of haemostasis and thrombosis. The journal is devoted to publishing significant developments worldwide in the field of blood coagulation, fibrinolysis, thrombosis, platelets and the kininogen-kinin system, as well as dealing with those aspects of blood rheology relevant to haemostasis and the effects of drugs on haemostatic components


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
