{"title":"A Case of Rafiq Syndrome (MAN1B1-CDG) in a Palestinian Child, With Brief Literature Review of 44 Cases.","authors":"Reema Iskafi, Bahaa AbuRahmeh, Roa'a Aljuneidi, Hidaya AlShweiki, Siraj Abdelnabi, Anas Abukhalaf, Bara' Maraqa","doi":"10.1177/23247096251313731","DOIUrl":null,"url":null,"abstract":"<p><p>Rafiq syndrome, MAN1B1-CDG, was described in 2010 and associated with genetic mutation in MAN1B1 gene in 2011. The disorder follows an autosomal recessive pattern of inheritance and typically presents with specific facial dysmorphism, intellectual disability, developmental delay, obesity, and hypotonia. The syndrome belongs to a group of metabolic disorders called Congenital Glycosylation Disorders (CGD). In this study, we discuss a 5-year-old male from Palestine who presented with developmental delay, hypotonia, characteristic facial dysmorphisms, impulsive behaviors, inability to speak, cryptorchidism, and other manifestations. This constellation of manifestations raised suspicion of a genetic disorder, prompting whole exome sequencing (WES), which revealed the presence of a homozygous likely pathogenic variant in the MAN1B1 gene (c.1976T>G)(p.Phe659Cys). We also reviewed all previously documented cases and compared the clinical features among them. After reviewing the family pedigree and its suspected cases, we found that the 2 most frequent features among them are intellectual disability and facial dysmorphism, whereas the least frequent one is truncal obesity. We discussed the importance of providing genetic counseling to parents of children with this and other rare, autosomal recessive disorders to prevent new cases from appearing.</p>","PeriodicalId":16198,"journal":{"name":"Journal of investigative medicine high impact case reports","volume":"13 ","pages":"23247096251313731"},"PeriodicalIF":0.8000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11755523/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of investigative medicine high impact case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/23247096251313731","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
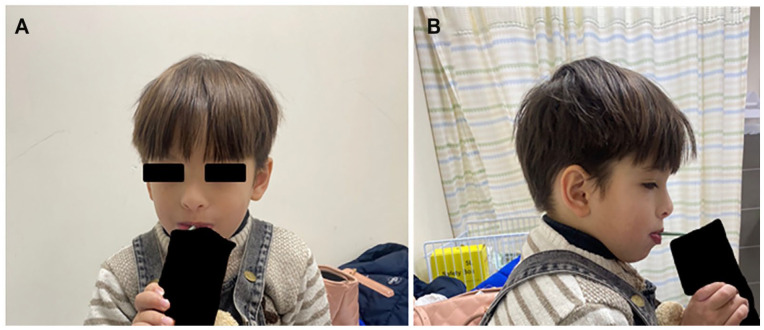
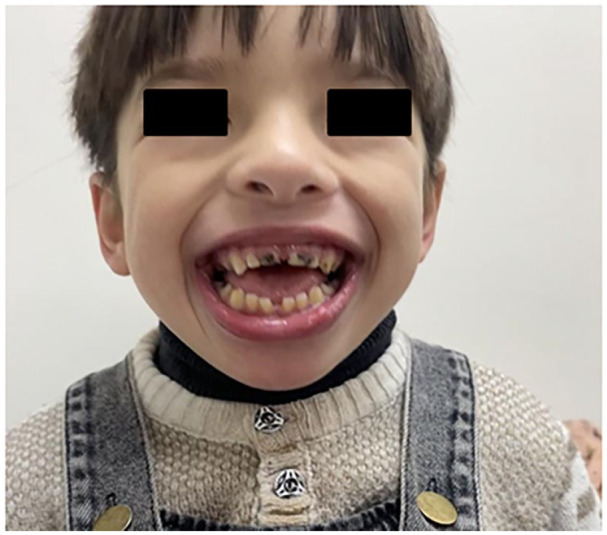
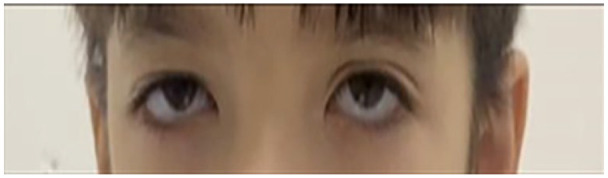
Rafiq syndrome, MAN1B1-CDG, was described in 2010 and associated with genetic mutation in MAN1B1 gene in 2011. The disorder follows an autosomal recessive pattern of inheritance and typically presents with specific facial dysmorphism, intellectual disability, developmental delay, obesity, and hypotonia. The syndrome belongs to a group of metabolic disorders called Congenital Glycosylation Disorders (CGD). In this study, we discuss a 5-year-old male from Palestine who presented with developmental delay, hypotonia, characteristic facial dysmorphisms, impulsive behaviors, inability to speak, cryptorchidism, and other manifestations. This constellation of manifestations raised suspicion of a genetic disorder, prompting whole exome sequencing (WES), which revealed the presence of a homozygous likely pathogenic variant in the MAN1B1 gene (c.1976T>G)(p.Phe659Cys). We also reviewed all previously documented cases and compared the clinical features among them. After reviewing the family pedigree and its suspected cases, we found that the 2 most frequent features among them are intellectual disability and facial dysmorphism, whereas the least frequent one is truncal obesity. We discussed the importance of providing genetic counseling to parents of children with this and other rare, autosomal recessive disorders to prevent new cases from appearing.
期刊介绍:
The AFMR is committed to enhancing the training and career development of our members and to furthering its mission to facilitate the conduct of research to improve medical care. Case reports represent an important avenue for trainees (interns, residents, and fellows) and early-stage faculty to demonstrate productive, scholarly activity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
