Ambiguous Genitalia Due to 3β-Hydroxysteroid Dehydrogenase Type 2 Deficiency: Clinical, Genetic, and Functional Characterization of Two Novel HSD3B2 Variants.
Jani Liimatta, Kay Sauter, Therina du Toit, André Schaller, Dagmar l'Allemand, Christa E Flück
{"title":"Ambiguous Genitalia Due to 3β-Hydroxysteroid Dehydrogenase Type 2 Deficiency: Clinical, Genetic, and Functional Characterization of Two Novel <i>HSD3B2</i> Variants.","authors":"Jani Liimatta, Kay Sauter, Therina du Toit, André Schaller, Dagmar l'Allemand, Christa E Flück","doi":"10.1210/jcemcr/luae245","DOIUrl":null,"url":null,"abstract":"<p><p>3β-Hydroxysteroid dehydrogenase 2 deficiency (3βHSD2D) is a rare form of congenital adrenal hyperplasia (CAH) with variable clinical presentation. We describe a 46, XY child with ambiguous genitalia and CAH without apparent adrenal insufficiency due to 2 novel heterozygous variants in the <i>HSD3B2</i> gene (c.779C > T/p.Pro260Leu and c.307 + 1G > A/p.Gly103Asp,fs29X). The disease-causing effect of the novel variants was assessed by genetic and functional studies informing on positive genotype-phenotype correlation. Sex registration was female, and no gender dysphoria has been noted until the present age of 7 years, but psychological assessments have been difficult with a concomitant diagnosis of autism spectrum disorder. Virilization that already progresses prepubertally through peripheral conversion of androgen precursors by 3β-hydroxysteroid dehydrogenase 1 will pose an increasing challenge during puberty.</p>","PeriodicalId":73540,"journal":{"name":"JCEM case reports","volume":"3 2","pages":"luae245"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11744041/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JCEM case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jcemcr/luae245","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
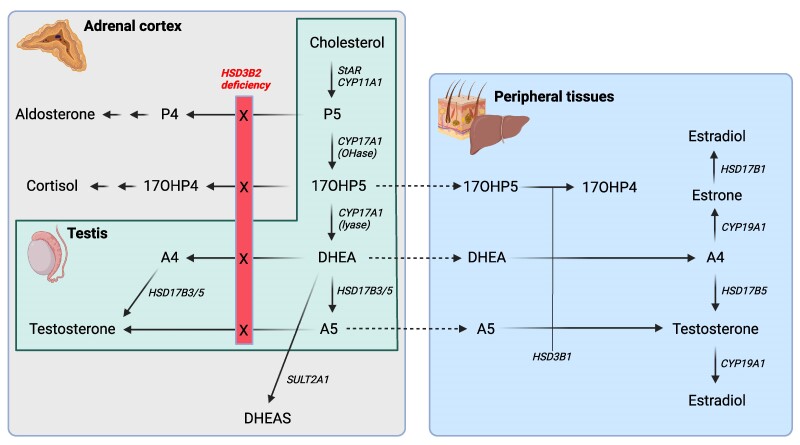
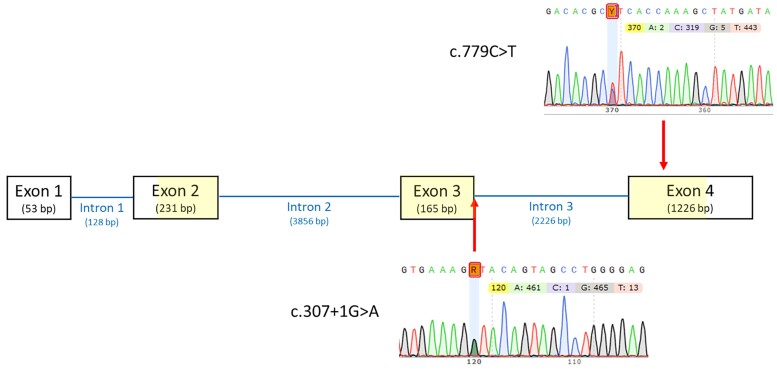
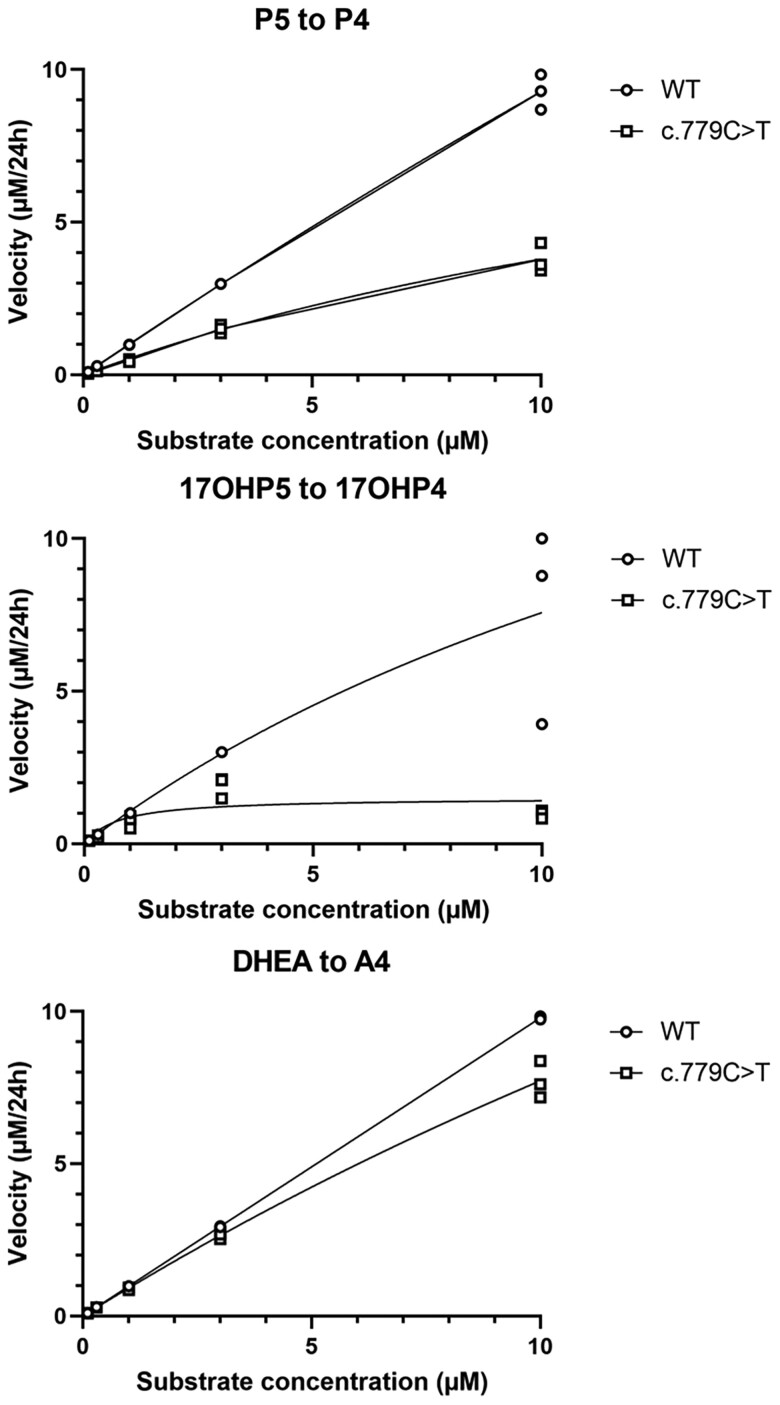
3β-Hydroxysteroid dehydrogenase 2 deficiency (3βHSD2D) is a rare form of congenital adrenal hyperplasia (CAH) with variable clinical presentation. We describe a 46, XY child with ambiguous genitalia and CAH without apparent adrenal insufficiency due to 2 novel heterozygous variants in the HSD3B2 gene (c.779C > T/p.Pro260Leu and c.307 + 1G > A/p.Gly103Asp,fs29X). The disease-causing effect of the novel variants was assessed by genetic and functional studies informing on positive genotype-phenotype correlation. Sex registration was female, and no gender dysphoria has been noted until the present age of 7 years, but psychological assessments have been difficult with a concomitant diagnosis of autism spectrum disorder. Virilization that already progresses prepubertally through peripheral conversion of androgen precursors by 3β-hydroxysteroid dehydrogenase 1 will pose an increasing challenge during puberty.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
