Laura Koponen, Minna Pekkinen, Jelmer Legebeke, Mari Muurinen, Salla Rusanen, Shabir Hussain, Fan Wang, Pasi I Nevalainen, Outi Mäkitie
{"title":"A deep intronic <i>PHEX</i> variant associated with X-linked hypophosphatemia in a Finnish family.","authors":"Laura Koponen, Minna Pekkinen, Jelmer Legebeke, Mari Muurinen, Salla Rusanen, Shabir Hussain, Fan Wang, Pasi I Nevalainen, Outi Mäkitie","doi":"10.1093/jbmrpl/ziae169","DOIUrl":null,"url":null,"abstract":"<p><p>Hypophosphatemic rickets is a rare bone disease characterized by short stature, bone deformities, impaired bone mineralization, and dental problems. Most commonly, hypophosphatemic rickets is caused by pathogenic variants in the X-chromosomal <i>PHEX</i> gene, but autosomal dominant and recessive forms also exist. We investigated a Finnish family in which the son (index, 29 yr) and mother (56 yr) had hypophosphatemia since childhood. Both patients had typical clinical, radiographic, and biochemical features of hypophosphatemic rickets, including a pathological fracture in the son. Gene panels and whole-exome sequencing did not reveal any pathogenic variants in the known hypophosphatemia genes. Therefore, we performed whole genome sequencing and identified a deep intronic variant (c.2147 + 1197A > G) in <i>PHEX</i>. Both the affected individuals, but none of the unaffected family members, had the same variant, as confirmed by Sanger sequencing. According to RT-PCR, whole transcriptomic data, and in silico analyses, the variant led to a new splice donor site in intron 21 and an 84 basepair pseudoexon between exons 21 and 22, likely leading to the synthesis of abnormal PHEX protein. Our study underscores the importance of intronic <i>PHEX</i> variants in X-linked hypophosphatemia (XLH). In patients with features of XLH but negative gene panel or whole-exome sequencing results, the combination of whole-genome sequencing and whole transcriptomics should be considered to detect possible deep intronic variants. The methodologies presented have the potential to be used more widely in other rare diseases.</p>","PeriodicalId":14611,"journal":{"name":"JBMR Plus","volume":"9 2","pages":"ziae169"},"PeriodicalIF":2.4000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11772523/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JBMR Plus","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/jbmrpl/ziae169","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
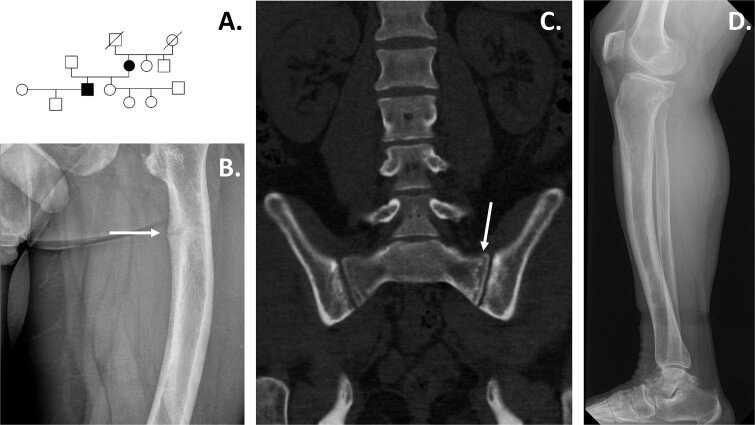
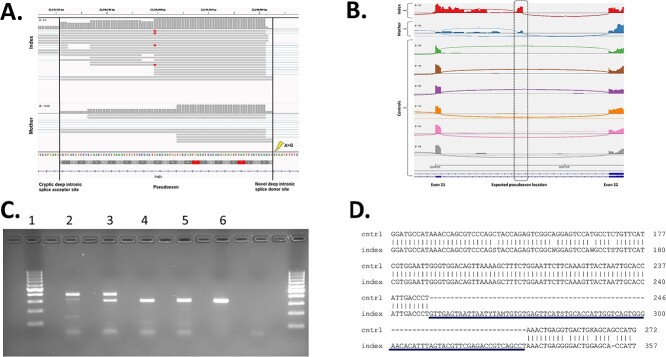
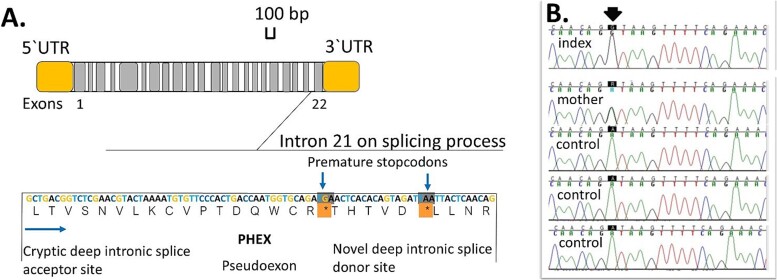
Hypophosphatemic rickets is a rare bone disease characterized by short stature, bone deformities, impaired bone mineralization, and dental problems. Most commonly, hypophosphatemic rickets is caused by pathogenic variants in the X-chromosomal PHEX gene, but autosomal dominant and recessive forms also exist. We investigated a Finnish family in which the son (index, 29 yr) and mother (56 yr) had hypophosphatemia since childhood. Both patients had typical clinical, radiographic, and biochemical features of hypophosphatemic rickets, including a pathological fracture in the son. Gene panels and whole-exome sequencing did not reveal any pathogenic variants in the known hypophosphatemia genes. Therefore, we performed whole genome sequencing and identified a deep intronic variant (c.2147 + 1197A > G) in PHEX. Both the affected individuals, but none of the unaffected family members, had the same variant, as confirmed by Sanger sequencing. According to RT-PCR, whole transcriptomic data, and in silico analyses, the variant led to a new splice donor site in intron 21 and an 84 basepair pseudoexon between exons 21 and 22, likely leading to the synthesis of abnormal PHEX protein. Our study underscores the importance of intronic PHEX variants in X-linked hypophosphatemia (XLH). In patients with features of XLH but negative gene panel or whole-exome sequencing results, the combination of whole-genome sequencing and whole transcriptomics should be considered to detect possible deep intronic variants. The methodologies presented have the potential to be used more widely in other rare diseases.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
