Yang Wang, Qian Wang, Xijun Wang, Jing Yang, Jun Jiang, Chuanyi Jia
{"title":"Accelerated Design of Dual-Metal-Site Catalysts via Machine-Learning Prediction.","authors":"Yang Wang, Qian Wang, Xijun Wang, Jing Yang, Jun Jiang, Chuanyi Jia","doi":"10.1021/acs.jpclett.5c00126","DOIUrl":null,"url":null,"abstract":"<p><p>Dual-metal site catalysts (DMSCs) supported on nitrogen-doped graphene have shown great potential in heterogeneous catalysis due to their unique properties and enhanced efficiency. However, the precise control and stabilization of metal dimers, particularly in oxygen activation reactions, present significant challenges in practical applications. In this study, we integrate high-throughput density functional theory calculations with machine learning techniques to predict and optimize the catalytic properties of DMSCs. Transfer learning is employed to enhance the model's generalization capability, successfully predicting catalytic performance across new metal combinations. Additionally, the application of the SISSO method enables the derivation of interpretable symbolic regression models, revealing critical correlations between electronic structure features and catalytic efficiency. This approach not only advances the understanding of dual-metal site catalysis but also provides a novel framework for the systematic design and optimization of highly efficient catalysts, with broad applicability in catalytic science.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":" ","pages":"1424-1431"},"PeriodicalIF":4.6000,"publicationDate":"2025-02-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.5c00126","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/31 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
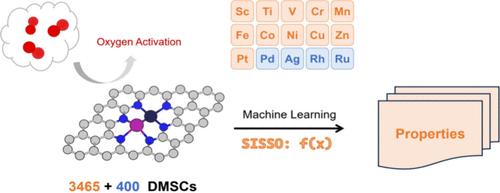
Dual-metal site catalysts (DMSCs) supported on nitrogen-doped graphene have shown great potential in heterogeneous catalysis due to their unique properties and enhanced efficiency. However, the precise control and stabilization of metal dimers, particularly in oxygen activation reactions, present significant challenges in practical applications. In this study, we integrate high-throughput density functional theory calculations with machine learning techniques to predict and optimize the catalytic properties of DMSCs. Transfer learning is employed to enhance the model's generalization capability, successfully predicting catalytic performance across new metal combinations. Additionally, the application of the SISSO method enables the derivation of interpretable symbolic regression models, revealing critical correlations between electronic structure features and catalytic efficiency. This approach not only advances the understanding of dual-metal site catalysis but also provides a novel framework for the systematic design and optimization of highly efficient catalysts, with broad applicability in catalytic science.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
