Juyoung Sung , Insung Kim , Minji Im , Yoon Ji Ahn , Sang-Mi Kim , Ja-Hyun Jang , Hyung-Doo Park , Tae Yeon Jeon , Kyung Rae Ko , Se-Jun Park , Jun Hwa Lee , Eun Young Kim , Chong Kun Cheon , Eungu Kang , Jung-eun Moon , Young Bae Sohn , Hsiang-Yu Lin , Chih-Kuang Chuang , Shuan-Pei Lin , Sung Yoon Cho
{"title":"Long-term outcomes of enzyme replacement therapy from a large cohort of Korean patients with mucopolysaccharidosis IVA (Morquio A syndrome)","authors":"Juyoung Sung , Insung Kim , Minji Im , Yoon Ji Ahn , Sang-Mi Kim , Ja-Hyun Jang , Hyung-Doo Park , Tae Yeon Jeon , Kyung Rae Ko , Se-Jun Park , Jun Hwa Lee , Eun Young Kim , Chong Kun Cheon , Eungu Kang , Jung-eun Moon , Young Bae Sohn , Hsiang-Yu Lin , Chih-Kuang Chuang , Shuan-Pei Lin , Sung Yoon Cho","doi":"10.1016/j.ymgmr.2025.101189","DOIUrl":null,"url":null,"abstract":"<div><div>Mucopolysaccharidosis (MPS) IVA (Morquio A syndrome) is an autosomal recessive lysosomal storage disorder caused by a mutation affecting the enzyme <em>N</em>-acetylgalactosamine-6-sulfatase (EC 3.1.6.4, GALNS). Enzyme replacement therapy (ERT) has been shown to improve physical performance, quality of life, and respiratory function in patients with MPS IVA; however, owing to the rarity of MPS IVA, data on Korean patient characteristics are limited. This retrospective study reports clinical, radiographic, biochemical, and molecular findings, and analyzes long-term clinical outcomes, from the largest cohort of Korean patients with MPS IVA in a single center. The analysis included 17 patients from 14 families (58.8 % females; median [range] age at diagnosis 5.2 [1.8–33.7] years). The majority of patients (64.7 %) were classified as having a severe phenotype, 23 % had an intermediate phenotype, and 11.8 % had an attenuated phenotype. Skeletal manifestations and radiologic abnormalities at initial diagnosis included gait abnormality (35.3 %), short stature (23.5 %), chest deformity (23.5 %), scoliosis (17.6 %), kyphosis (11.8 %), dysmorphic face (6 %), hip pain (6 %), and leg deformity (6 %). Twelve different <em>GALNS</em> mutations were identified. Patients received ERT for a median (range) 7.4 years (3.0–12.1). Twelve patients reached final adult height, and all patients with the severe/intermediate phenotype had short stature (<3rd percentile). Hemiepiphysiodesis was the most common surgical intervention among patients with the severe/intermediate phenotype. Drug-related adverse events (urticaria, rash, and anaphylaxis) were reported in four patients but were managed with antihistamines or desensitization. At follow-up, patients experienced improvements in functional independence measure score, ejection fraction, and the 6-min walk test compared with the pre-treatment baseline. This study provides real-world evidence for long-term stabilization of functional independence, endurance, and respiratory function among patients <strong>with MPS IVA</strong> treated with ERT, with no new safety concerns identified.</div></div>","PeriodicalId":18814,"journal":{"name":"Molecular Genetics and Metabolism Reports","volume":"42 ","pages":"Article 101189"},"PeriodicalIF":1.9000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11783393/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics and Metabolism Reports","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2214426925000047","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/15 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
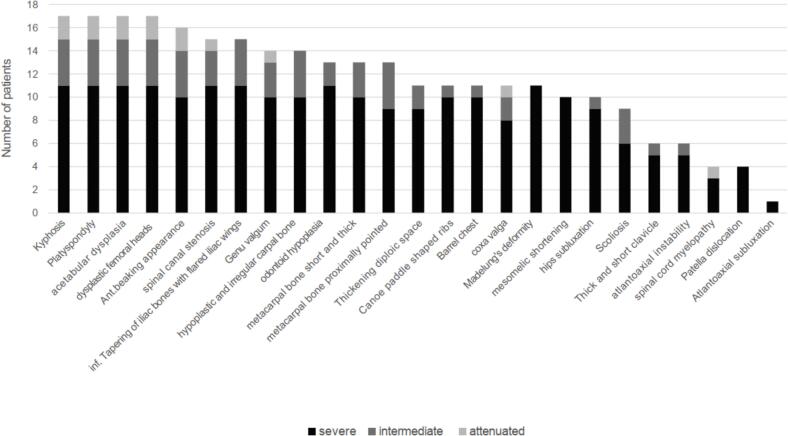
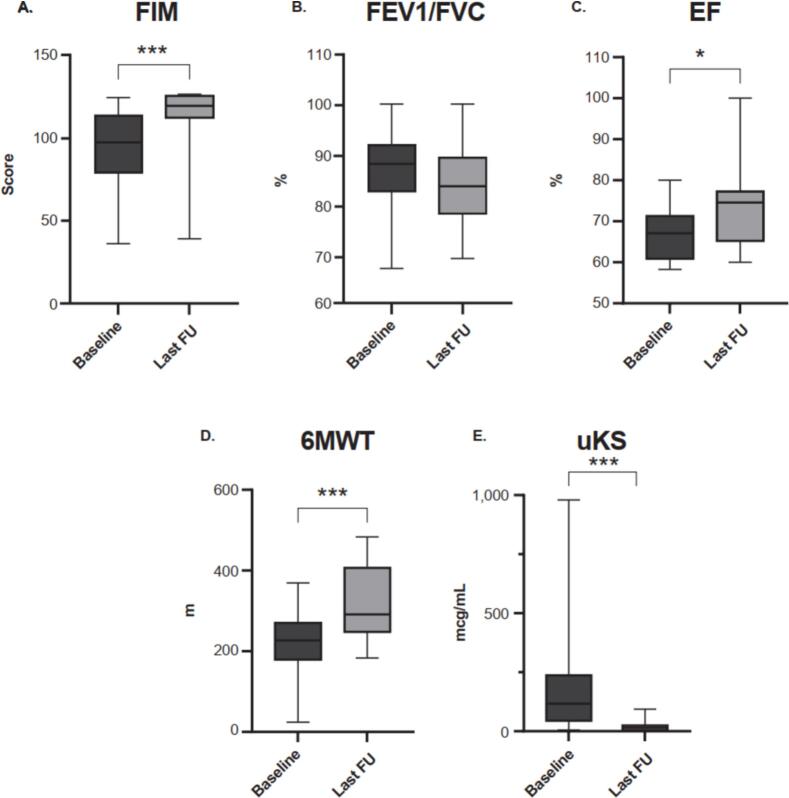
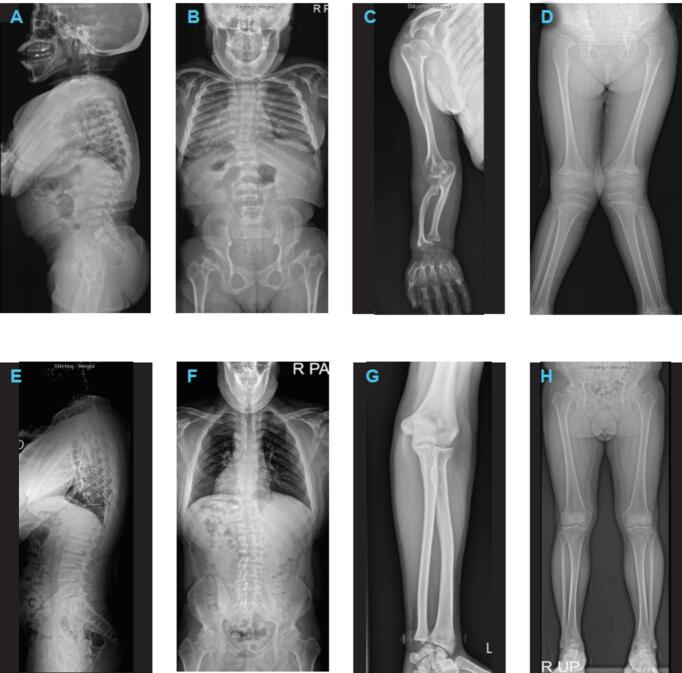
Mucopolysaccharidosis (MPS) IVA (Morquio A syndrome) is an autosomal recessive lysosomal storage disorder caused by a mutation affecting the enzyme N-acetylgalactosamine-6-sulfatase (EC 3.1.6.4, GALNS). Enzyme replacement therapy (ERT) has been shown to improve physical performance, quality of life, and respiratory function in patients with MPS IVA; however, owing to the rarity of MPS IVA, data on Korean patient characteristics are limited. This retrospective study reports clinical, radiographic, biochemical, and molecular findings, and analyzes long-term clinical outcomes, from the largest cohort of Korean patients with MPS IVA in a single center. The analysis included 17 patients from 14 families (58.8 % females; median [range] age at diagnosis 5.2 [1.8–33.7] years). The majority of patients (64.7 %) were classified as having a severe phenotype, 23 % had an intermediate phenotype, and 11.8 % had an attenuated phenotype. Skeletal manifestations and radiologic abnormalities at initial diagnosis included gait abnormality (35.3 %), short stature (23.5 %), chest deformity (23.5 %), scoliosis (17.6 %), kyphosis (11.8 %), dysmorphic face (6 %), hip pain (6 %), and leg deformity (6 %). Twelve different GALNS mutations were identified. Patients received ERT for a median (range) 7.4 years (3.0–12.1). Twelve patients reached final adult height, and all patients with the severe/intermediate phenotype had short stature (<3rd percentile). Hemiepiphysiodesis was the most common surgical intervention among patients with the severe/intermediate phenotype. Drug-related adverse events (urticaria, rash, and anaphylaxis) were reported in four patients but were managed with antihistamines or desensitization. At follow-up, patients experienced improvements in functional independence measure score, ejection fraction, and the 6-min walk test compared with the pre-treatment baseline. This study provides real-world evidence for long-term stabilization of functional independence, endurance, and respiratory function among patients with MPS IVA treated with ERT, with no new safety concerns identified.
期刊介绍:
Molecular Genetics and Metabolism Reports is an open access journal that publishes molecular and metabolic reports describing investigations that use the tools of biochemistry and molecular biology for studies of normal and diseased states. In addition to original research articles, sequence reports, brief communication reports and letters to the editor are considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
