{"title":"Genetics in Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency and Clinical Implications.","authors":"Paola Concolino, Henrik Falhammar","doi":"10.1210/jendso/bvaf018","DOIUrl":null,"url":null,"abstract":"<p><p>Of all congenital adrenal hyperplasia (CAH), 95% to 99% is 21-hydroxylase deficiency (21OHD), an autosomal recessive disease. 21OHD is due to an insufficiency of 21-hydroxylase enzyme, which is encoded by the <i>CYP21A2</i> gene and involved in cortisol and aldosterone production. The clinical presentation differs widely from severe classic to mild nonclassic CAH. 21OHD represents one of the most complex and at the same time intriguing topics in human genetics and its molecular diagnosis involves ongoing challenges. To provide a meticulous presentation of the topic, we searched the past and present literature, including original articles and reviews from PubMed, ScienceDirect, Web of Science, Embase, and Scopus, using search terms for genetics of 21OHD, 21OHD variants, molecular diagnosis of 21OHD, and 21OHD genetic testing. We offer a comprehensive review focusing on recent developments, new concepts, and conclusions.</p>","PeriodicalId":17334,"journal":{"name":"Journal of the Endocrine Society","volume":"9 3","pages":"bvaf018"},"PeriodicalIF":3.1000,"publicationDate":"2025-01-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11795198/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the Endocrine Society","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jendso/bvaf018","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/4 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
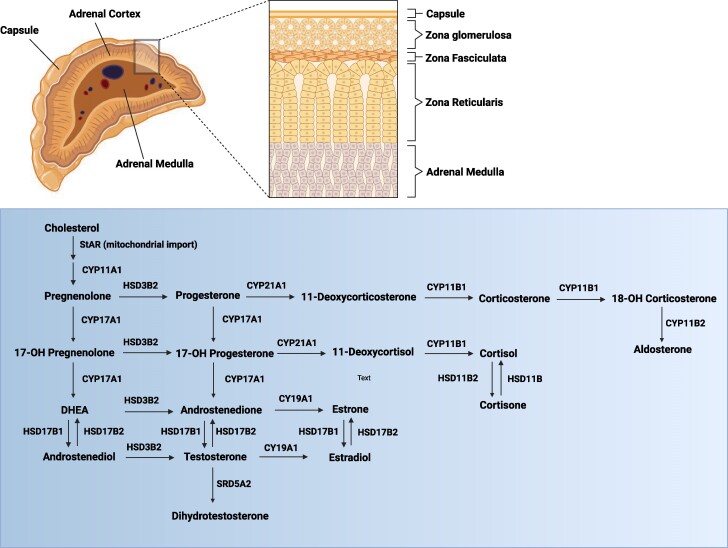
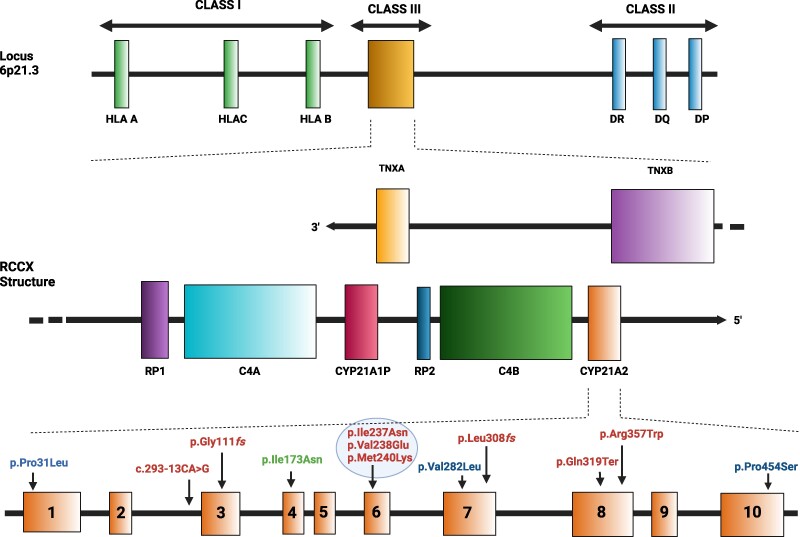
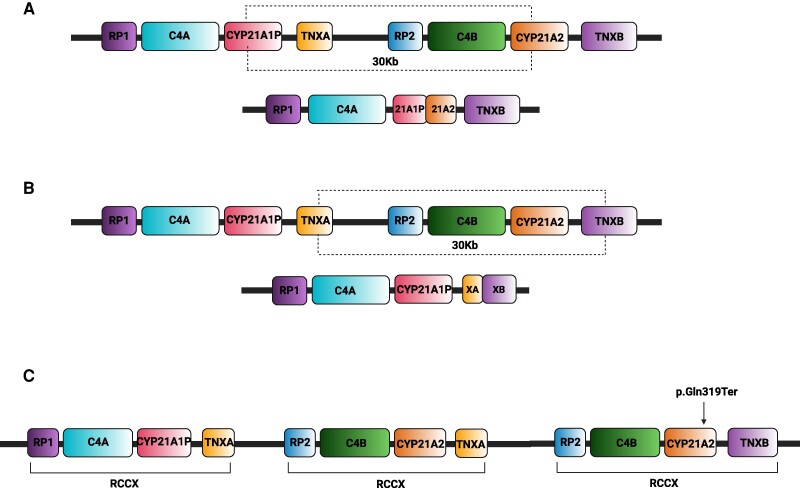
Of all congenital adrenal hyperplasia (CAH), 95% to 99% is 21-hydroxylase deficiency (21OHD), an autosomal recessive disease. 21OHD is due to an insufficiency of 21-hydroxylase enzyme, which is encoded by the CYP21A2 gene and involved in cortisol and aldosterone production. The clinical presentation differs widely from severe classic to mild nonclassic CAH. 21OHD represents one of the most complex and at the same time intriguing topics in human genetics and its molecular diagnosis involves ongoing challenges. To provide a meticulous presentation of the topic, we searched the past and present literature, including original articles and reviews from PubMed, ScienceDirect, Web of Science, Embase, and Scopus, using search terms for genetics of 21OHD, 21OHD variants, molecular diagnosis of 21OHD, and 21OHD genetic testing. We offer a comprehensive review focusing on recent developments, new concepts, and conclusions.
在所有先天性肾上腺增生症(CAH)中,95%至99%为21-羟化酶缺乏症(21OHD),这是一种常染色体隐性遗传病。21OHD是由于21-羟化酶不足,该酶由CYP21A2基因编码,参与皮质醇和醛固酮的产生。临床表现从严重的经典到轻度的非经典CAH差别很大。21OHD是人类遗传学中最复杂,同时也是最有趣的话题之一,其分子诊断涉及持续的挑战。为了提供一个细致的主题展示,我们检索了过去和现在的文献,包括来自PubMed, ScienceDirect, Web of Science, Embase和Scopus的原创文章和评论,使用21OHD遗传学,21OHD变体,21OHD分子诊断和21OHD基因检测的搜索词。我们提供了一个全面的审查,重点是最近的发展,新的概念和结论。



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
