{"title":"A Novel Truncating Variant in Sandestig-Stefanova Syndrome with Hydrocephalus.","authors":"Gülnihal Bulut, Gözde Tutku Turgut, Güven Toksoy, Umut Altunoğlu, Ayça Dilruba Aslanger, Zehra Oya Uyguner, Birsen Karaman","doi":"10.1159/000540314","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Sandestig-Stefanova syndrome (MIM:618804) is characterized by pre- and postnatal microcephaly, trigonocephaly, bilateral congenital cataracts, microphthalmia, cleft lip and palate or high-arched palate, camptodactyly, rocker-bottom feet, heart anomalies, periventricular white matter loss, thin corpus callosum, and delayed myelination. Bi-allelic loss-of-function variants in the <i>Nucleoporin 188 (NUP188)</i> (MIM:615587) gene are implicated in the etiology.</p><p><strong>Case presentation: </strong>Our patient, born to consanguineous parents, presented with tetralogy of Fallot, bilateral congenital cataracts, hydrocephalus, a bifid uvula, a right pelvic kidney, hepatomegaly, facial feature findings, and a history of a similarly affected ex-sibling. Whole exome sequence analysis in the index case revealed a novel homozygous variant NM_015354.2: c.124C>T/p.(Arg42Ter) in the <i>NUP188</i> gene.</p><p><strong>Conclusion: </strong>This study describes a new patient with Sandestig-Stefanova syndrome harboring a novel pathogenic variant in the <i>NUP188</i> gene.</p>","PeriodicalId":48566,"journal":{"name":"Molecular Syndromology","volume":"16 1","pages":"69-76"},"PeriodicalIF":0.9000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11793897/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Syndromology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1159/000540314","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/13 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Sandestig-Stefanova syndrome (MIM:618804) is characterized by pre- and postnatal microcephaly, trigonocephaly, bilateral congenital cataracts, microphthalmia, cleft lip and palate or high-arched palate, camptodactyly, rocker-bottom feet, heart anomalies, periventricular white matter loss, thin corpus callosum, and delayed myelination. Bi-allelic loss-of-function variants in the Nucleoporin 188 (NUP188) (MIM:615587) gene are implicated in the etiology.
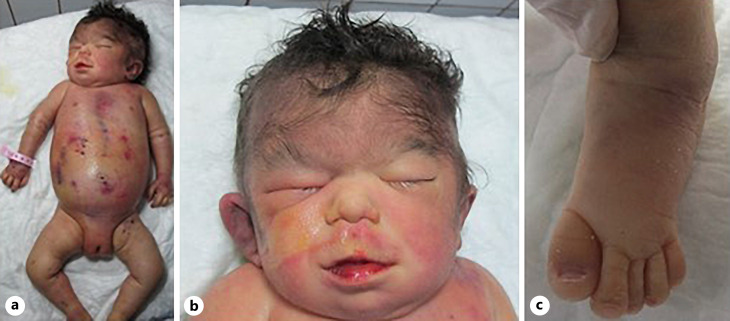
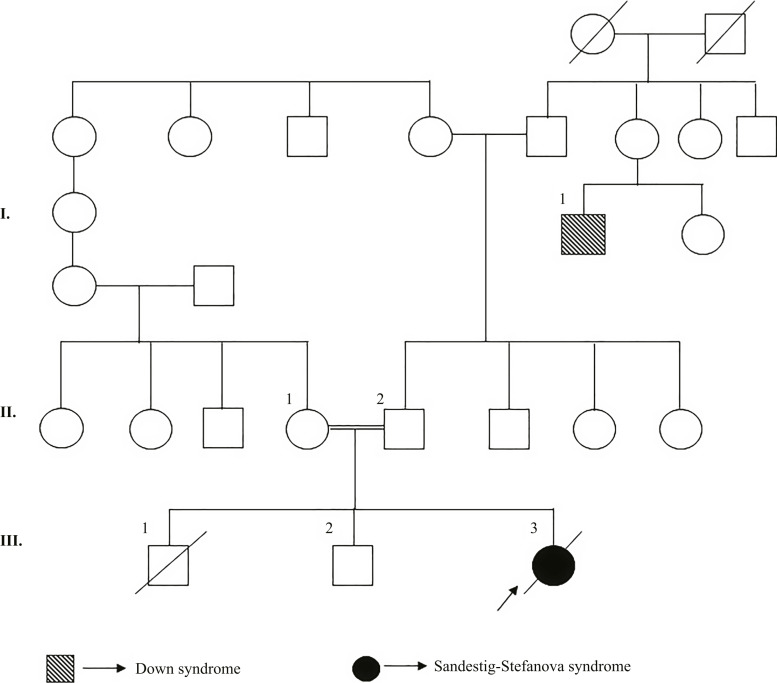
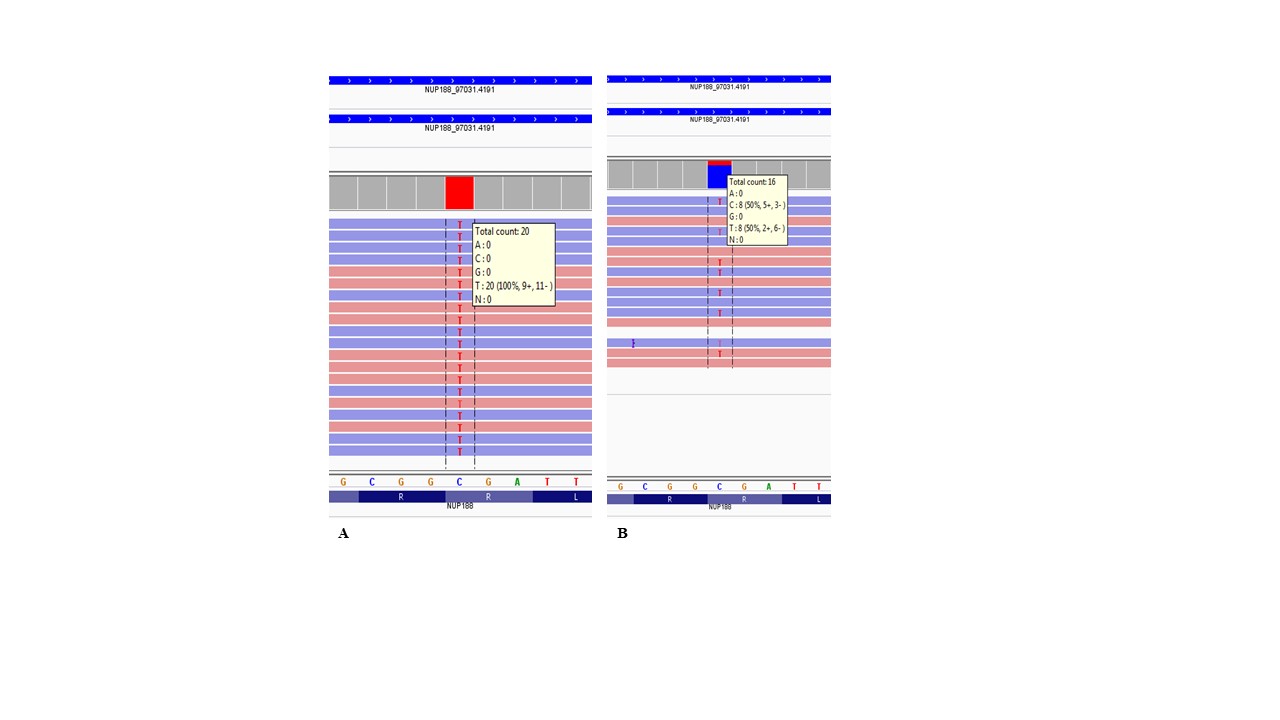
Case presentation: Our patient, born to consanguineous parents, presented with tetralogy of Fallot, bilateral congenital cataracts, hydrocephalus, a bifid uvula, a right pelvic kidney, hepatomegaly, facial feature findings, and a history of a similarly affected ex-sibling. Whole exome sequence analysis in the index case revealed a novel homozygous variant NM_015354.2: c.124C>T/p.(Arg42Ter) in the NUP188 gene.
Conclusion: This study describes a new patient with Sandestig-Stefanova syndrome harboring a novel pathogenic variant in the NUP188 gene.
期刊介绍:
''Molecular Syndromology'' publishes high-quality research articles, short reports and reviews on common and rare genetic syndromes, aiming to increase clinical understanding through molecular insights. Topics of particular interest are the molecular basis of genetic syndromes, genotype-phenotype correlation, natural history, strategies in disease management and novel therapeutic approaches based on molecular findings. Research on model systems is also welcome, especially when it is obviously relevant to human genetics. With high-quality reviews on current topics the journal aims to facilitate translation of research findings to a clinical setting while also stimulating further research on clinically relevant questions. The journal targets not only medical geneticists and basic biomedical researchers, but also clinicians dealing with genetic syndromes. With four Associate Editors from three continents and a broad international Editorial Board the journal welcomes submissions covering the latest research from around the world.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
