Computational Study of the Transfer Hydrogenation of Furfural to Furfuryl Alcohol in the UiO-66 Metal–Organic Frameworks: Mechanistic Insights and Predictive Descriptors for Transition States and Activation Energies
{"title":"Computational Study of the Transfer Hydrogenation of Furfural to Furfuryl Alcohol in the UiO-66 Metal–Organic Frameworks: Mechanistic Insights and Predictive Descriptors for Transition States and Activation Energies","authors":"Thana Maihom, Bundet Boekfa, Sasiwadee Boonya-Udtayan, Pradudnet Ketwong, Chularat Wattanakit, Michael Probst, Jumras Limtrakul","doi":"10.1021/acs.jpcc.4c07926","DOIUrl":null,"url":null,"abstract":"The catalytic transfer hydrogenation (CTH) of carbonyls in biobased chemicals is an essential route for converting biomass into more valuable chemicals and fuels. In this work, we investigate the CTH of furfural to furfuryl alcohol using isopropanol as the hydrogen source over UiO-66 metal–organic frameworks by means of density functional calculations. Two main reaction pathways differing in the formation of isopropoxide are analyzed. The pathway without isopropoxide formation is found to be favored over the one where isopropoxide is formed due to smaller activation barriers in the rate-limiting step and a subsequently higher rate constant. The acid strength of UiO-66 can be tuned by partial substitution of tetravalent metals, with the barriers appearing in the order Hf < Zr < Ce < Ti. Therefore, the catalytic activity of the Hf-substituted zeolite is the highest. In turn, modifying the group connecting the Zr sites of the MOF with a −CH<sub>3</sub> group reduces the barrier. Regression analysis shows that both the partial charge of the tetravalent metal site bound to furfural and the NH<sub>3</sub> adsorption energies serve as good descriptors for predicting the transition state energy, while the activation barrier can be reasonably approximated by linear functions of the partial charge of the tetravalent metal site alone. By means of the sure-independence screening and sparsifying operator (SISSO) method, we can identify the best nonlinear combination of DFT-based descriptors. We find that the combinations of descriptors have excellent predictions that can, in many cases, eliminate the need for time-intensive quantum chemical reaction pathway calculations.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"23 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2025-02-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c07926","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
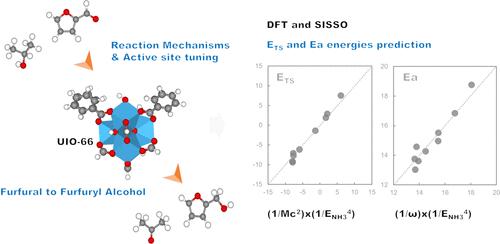
The catalytic transfer hydrogenation (CTH) of carbonyls in biobased chemicals is an essential route for converting biomass into more valuable chemicals and fuels. In this work, we investigate the CTH of furfural to furfuryl alcohol using isopropanol as the hydrogen source over UiO-66 metal–organic frameworks by means of density functional calculations. Two main reaction pathways differing in the formation of isopropoxide are analyzed. The pathway without isopropoxide formation is found to be favored over the one where isopropoxide is formed due to smaller activation barriers in the rate-limiting step and a subsequently higher rate constant. The acid strength of UiO-66 can be tuned by partial substitution of tetravalent metals, with the barriers appearing in the order Hf < Zr < Ce < Ti. Therefore, the catalytic activity of the Hf-substituted zeolite is the highest. In turn, modifying the group connecting the Zr sites of the MOF with a −CH3 group reduces the barrier. Regression analysis shows that both the partial charge of the tetravalent metal site bound to furfural and the NH3 adsorption energies serve as good descriptors for predicting the transition state energy, while the activation barrier can be reasonably approximated by linear functions of the partial charge of the tetravalent metal site alone. By means of the sure-independence screening and sparsifying operator (SISSO) method, we can identify the best nonlinear combination of DFT-based descriptors. We find that the combinations of descriptors have excellent predictions that can, in many cases, eliminate the need for time-intensive quantum chemical reaction pathway calculations.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
