A novel approach to analyzing the evolution of SARS-CoV-2 based on visualization and clustering of large genetic data compactly represented in operative memory.
{"title":"A novel approach to analyzing the evolution of SARS-CoV-2 based on visualization and clustering of large genetic data compactly represented in operative memory.","authors":"A Yu Palyanov, N V Palyanova","doi":"10.18699/vjgb-24-92","DOIUrl":null,"url":null,"abstract":"<p><p>SARS-CoV-2 is a virus for which an outstanding number of genome variants were collected, sequenced and stored from sources all around the world. Raw data in FASTA format include 16.8 million genomes, each ≈29,900 nt (nucleotides), with a total size of ≈500 ∙ 109 nt, or 465 Gb. We suggest an approach to data representation and organization, with which all this can be stored losslessly in the operative memory (RAM) of a common PC. Moreover, just ≈330 Mb will be enough. Aligning all genomes versus the initial Wuhan-Hu-1 reference sequence allows each to be represented as a data structure containing lists of point mutations, deletions and insertions. Our implementation of such data representation resulted in a 1:1500 compression ratio (for comparison, compression of the same data with the popular WinRAR archiver gives only 1:62) and fast access to genomes (and their metadata) and comparisons between different genome variants. With this approach implemented as a C++ program, we performed an analysis of various properties of the set of SARS-CoV-2 genomes available in NCBI Genbank (within a period from 24.12.2019 to 24.06.2024). We calculated the distribution of the number of genomes with undetermined nucleotides, 'N's, vs the number of such nucleotides in them, the number of unique genomes and clusters of identical genomes, and the distribution of clusters by size (the number of identical genomes) and duration (the time interval between each cluster's first and last genome). Finally, the evolution of distributions of the number of changes (editing distance between each genome and reference sequence) caused by substitutions, deletions and insertions was visualized as 3D surfaces, which clearly show the process of viral evolution over 4.5 years, with a time step = 1 week. It is in good correspondence with phylogenetic trees (usually based on 3-4 thousand of genome variant representatives), but is built over millions of genomes, shows more details and is independent of the type of lineage/clade classification.</p>","PeriodicalId":44339,"journal":{"name":"Vavilovskii Zhurnal Genetiki i Selektsii","volume":"28 8","pages":"843-853"},"PeriodicalIF":1.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11811502/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Vavilovskii Zhurnal Genetiki i Selektsii","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.18699/vjgb-24-92","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"AGRICULTURE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
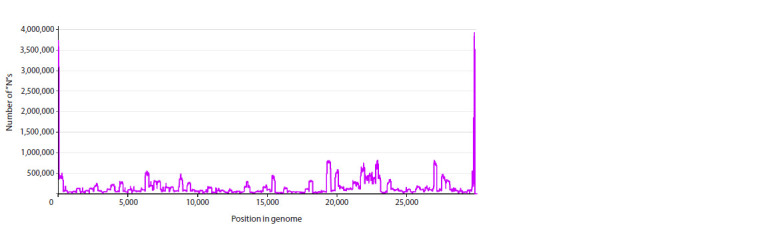
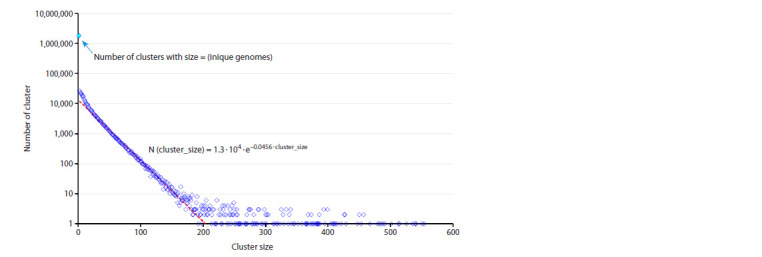
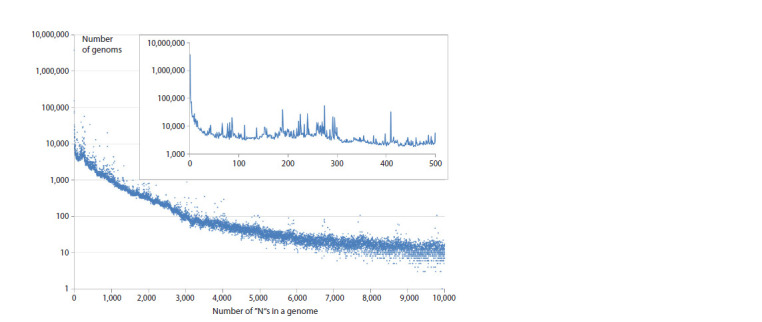
SARS-CoV-2 is a virus for which an outstanding number of genome variants were collected, sequenced and stored from sources all around the world. Raw data in FASTA format include 16.8 million genomes, each ≈29,900 nt (nucleotides), with a total size of ≈500 ∙ 109 nt, or 465 Gb. We suggest an approach to data representation and organization, with which all this can be stored losslessly in the operative memory (RAM) of a common PC. Moreover, just ≈330 Mb will be enough. Aligning all genomes versus the initial Wuhan-Hu-1 reference sequence allows each to be represented as a data structure containing lists of point mutations, deletions and insertions. Our implementation of such data representation resulted in a 1:1500 compression ratio (for comparison, compression of the same data with the popular WinRAR archiver gives only 1:62) and fast access to genomes (and their metadata) and comparisons between different genome variants. With this approach implemented as a C++ program, we performed an analysis of various properties of the set of SARS-CoV-2 genomes available in NCBI Genbank (within a period from 24.12.2019 to 24.06.2024). We calculated the distribution of the number of genomes with undetermined nucleotides, 'N's, vs the number of such nucleotides in them, the number of unique genomes and clusters of identical genomes, and the distribution of clusters by size (the number of identical genomes) and duration (the time interval between each cluster's first and last genome). Finally, the evolution of distributions of the number of changes (editing distance between each genome and reference sequence) caused by substitutions, deletions and insertions was visualized as 3D surfaces, which clearly show the process of viral evolution over 4.5 years, with a time step = 1 week. It is in good correspondence with phylogenetic trees (usually based on 3-4 thousand of genome variant representatives), but is built over millions of genomes, shows more details and is independent of the type of lineage/clade classification.
期刊介绍:
The "Vavilov Journal of genetics and breeding" publishes original research and review articles in all key areas of modern plant, animal and human genetics, genomics, bioinformatics and biotechnology. One of the main objectives of the journal is integration of theoretical and applied research in the field of genetics. Special attention is paid to the most topical areas in modern genetics dealing with global concerns such as food security and human health.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
