{"title":"Predicting and interpreting EPR spectra of POPC lipid bilayers with transmembrane α-helical peptides from all-atom molecular dynamics simulations†","authors":"Andrea Catte and Vasily S. Oganesyan","doi":"10.1039/D4CP04802D","DOIUrl":null,"url":null,"abstract":"<p >This study reports a large-scale all-atom MD simulation of POPC lipid bilayers in the presence of different concentrations of the transmembrane peptide acetyl-K<small><sub>2</sub></small>(LA)<small><sub>12</sub></small>K<small><sub>2</sub></small>-amide ((LA)<small><sub>12</sub></small>) and doped with 5-PC paramagnetic spin probes used in EPR studies. We apply a combined MD-EPR simulation methodology for the prediction of EPR spectra directly and completely from MD trajectories. This approach serves three major purposes. Firstly, comparing predicted EPR spectra with experimental ones, which are highly sensitive to motions, provides an ultimate test bed for the force fields currently employed for modeling lipid bilayer systems with embedded proteins or peptides. Secondly, simulations of EPR spectra directly from the atomistic MD models simplify the interpretation of the EPR line shapes and their changes induced by the presence of peptides in the lipid bilayer. These changes are directly linked to the dynamics and order of spin probes and POPC host molecules. Lastly and importantly, we demonstrate how the MD-EPR methodology can be employed to test the validity and limitations of the widely used approach for the estimation of the order parameter of lipids directly from the EPR experimental line shapes.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 9","pages":" 4775-4784"},"PeriodicalIF":2.9000,"publicationDate":"2025-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04802d?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04802d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
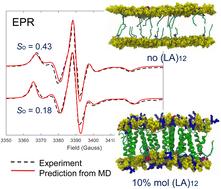
This study reports a large-scale all-atom MD simulation of POPC lipid bilayers in the presence of different concentrations of the transmembrane peptide acetyl-K2(LA)12K2-amide ((LA)12) and doped with 5-PC paramagnetic spin probes used in EPR studies. We apply a combined MD-EPR simulation methodology for the prediction of EPR spectra directly and completely from MD trajectories. This approach serves three major purposes. Firstly, comparing predicted EPR spectra with experimental ones, which are highly sensitive to motions, provides an ultimate test bed for the force fields currently employed for modeling lipid bilayer systems with embedded proteins or peptides. Secondly, simulations of EPR spectra directly from the atomistic MD models simplify the interpretation of the EPR line shapes and their changes induced by the presence of peptides in the lipid bilayer. These changes are directly linked to the dynamics and order of spin probes and POPC host molecules. Lastly and importantly, we demonstrate how the MD-EPR methodology can be employed to test the validity and limitations of the widely used approach for the estimation of the order parameter of lipids directly from the EPR experimental line shapes.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
