Anna Annunziata, Gerardo Langella, Rosa Cauteruccio, Luigi Fiorentino, Giuseppe Fiorentino
{"title":"Severe progressive respiratory involvement requiring ventilator support in autosomal recessive Bethlem myopathy. A case report.","authors":"Anna Annunziata, Gerardo Langella, Rosa Cauteruccio, Luigi Fiorentino, Giuseppe Fiorentino","doi":"10.36185/2532-1900-654","DOIUrl":null,"url":null,"abstract":"<p><p>Bethlem myopathy (BM) was first described in 1976 by Bethlem and van Wijngaarden in patients who presented a myopathy characterized by slowly progressive muscle weakness and typical flexion contractures of the long finger flexors, wrists, elbows, pectoralis muscles and ankles. Patients with Bethlem myopathy usually become symptomatic during the first or second decade of life. The condition is in most cases slowly progressive and more than two thirds of patients over 50 years of age may require aids for ambulation. Inheritance is usually autosomal dominant. However, patients with autosomal recessive (AR) BM have been recently reported in Literature. Cardiac involvement is usually absent. Respiratory muscle involvement necessitating nocturnal respiratory support is rarely reported in association with severe weakness later in life.</p><p><p>We describe a further case of ARBM in a 52-year-old man who presented a slowly progressive myopathy but developed a severe progressive respiratory involvement requiring ventilatory support.</p>","PeriodicalId":93851,"journal":{"name":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","volume":"43 4","pages":"149-152"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11978421/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-654","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
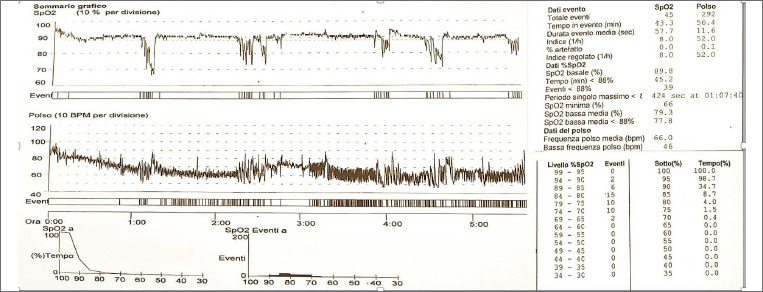
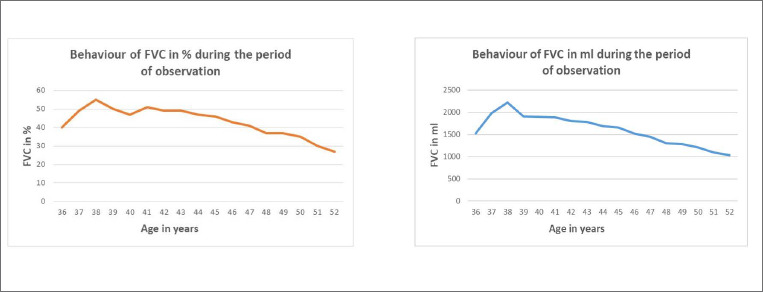
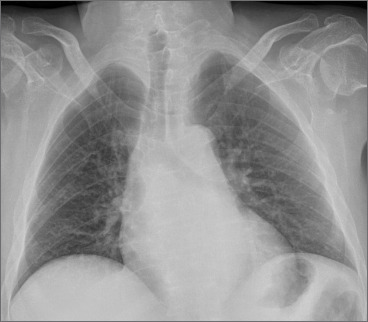
Bethlem myopathy (BM) was first described in 1976 by Bethlem and van Wijngaarden in patients who presented a myopathy characterized by slowly progressive muscle weakness and typical flexion contractures of the long finger flexors, wrists, elbows, pectoralis muscles and ankles. Patients with Bethlem myopathy usually become symptomatic during the first or second decade of life. The condition is in most cases slowly progressive and more than two thirds of patients over 50 years of age may require aids for ambulation. Inheritance is usually autosomal dominant. However, patients with autosomal recessive (AR) BM have been recently reported in Literature. Cardiac involvement is usually absent. Respiratory muscle involvement necessitating nocturnal respiratory support is rarely reported in association with severe weakness later in life.
We describe a further case of ARBM in a 52-year-old man who presented a slowly progressive myopathy but developed a severe progressive respiratory involvement requiring ventilatory support.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
