TP53 Mutations Detected by NGS Are a Major Clinical Risk Factor for Stratifying Mantle Cell Lymphoma
Grégory Lazarian, Lotfi Chemali, Merieme Bensalah, Carla Zindel, Valérie Lefebvre, Catherine Thieblemont, Antoine Martin, Giulia Tueur, Rémi Letestu, Carole Fleury, Vincent Leymarie, Valérie Vidal, Audrey Bidet, Elsa Maitre, Florence Cymbalista, Vincent Levy, Thierry Soussi, Fanny Baran-Marszak
下载PDF
{"title":"TP53 Mutations Detected by NGS Are a Major Clinical Risk Factor for Stratifying Mantle Cell Lymphoma","authors":"Grégory Lazarian, Lotfi Chemali, Merieme Bensalah, Carla Zindel, Valérie Lefebvre, Catherine Thieblemont, Antoine Martin, Giulia Tueur, Rémi Letestu, Carole Fleury, Vincent Leymarie, Valérie Vidal, Audrey Bidet, Elsa Maitre, Florence Cymbalista, Vincent Levy, Thierry Soussi, Fanny Baran-Marszak","doi":"10.1002/ajh.27650","DOIUrl":null,"url":null,"abstract":"<p>\n \n <figure>\n <div><picture>\n <source></source></picture><p></p>\n </div>\n </figure>\n </p>","PeriodicalId":7724,"journal":{"name":"American Journal of Hematology","volume":"100 5","pages":"933-936"},"PeriodicalIF":9.9000,"publicationDate":"2025-03-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajh.27650","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Hematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajh.27650","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
引用
批量引用
NGS检测TP53突变是分层套细胞淋巴瘤的主要临床危险因素
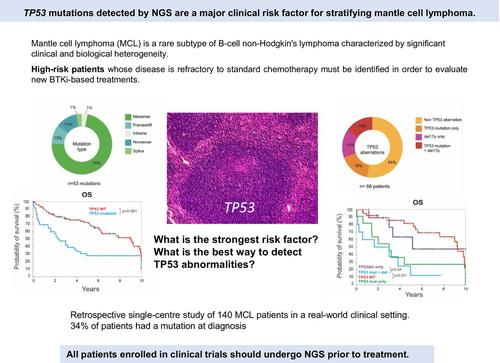
套细胞淋巴瘤(MCL)是一种罕见的b细胞非霍奇金淋巴瘤亚型,具有显著的临床和生物学异质性。最近,Bruton的酪氨酸激酶抑制剂(BTKi)联合化疗免疫疗法(CIT)联合或不联合自体干细胞移植(ASCT)在一线临床试验中显示出良好的效果。因此,有必要确定疾病对标准化疗难治性的高危患者,以评估新的基于btki的治疗方法。目前的预后因素包括套细胞淋巴瘤国际预后指数(MIPI)、组织学和细胞学特征、Ki-67水平升高和其他遗传畸变。TP53突变始终与不良预后相关,与早期疾病进展和死亡表现出强烈且独立的相关性,特别是在接受常规强化CIT以及r -苯达莫司汀、来那度胺或BTKi单独治疗的患者中。因此,修改治疗算法以考虑一线和复发MCL患者的TP53状态,是优化患者特异性管理的重要一步。一个主要的挑战是提高临床试验中对tp53突变患者的检测,以防止对突变患者的错误分类和对药物疗效的不准确评估。目前,并非所有MCL患者都常规进行TP53测序,临床试验采用了各种灵敏度和准确性不同的检测方法来识别TP53异常,导致结果不一致[3]。TP53评估通常依赖于活检组织的免疫组织化学分析,该分析可以预测错义突变,但无法检测TP53零突变。只有新一代测序(NGS)才能在各种样品类型[4]中以高灵敏度准确检测导致突变蛋白表达(如错义突变)或P53表达缺失(如无义突变、帧移和剪接位点改变)的全谱突变。NGS还能够检测反映克隆异质性的共存突变,并根据变异等位基因频率(VAF)对突变TP53克隆的大小提供可靠的估计,特别是对于具有高肿瘤细胞纯度的样品。然而,目前还没有关于突变类型或克隆异质性对患者生存影响的数据。为了评估突变的频率和类型及其对治疗后生存的影响,我们在现实世界的临床环境中对1986年至2023年间通过流式细胞术诊断为循环肿瘤细胞的140例MCL患者进行了一项回顾性、单中心研究。所有患者均来自阿维森纳和圣路易斯医院,并提供知情同意;这些患者被纳入“b细胞淋巴增生性疾病”队列(DC 2009 936)。使用NGS进行TP53测序,包括整个编码序列和外显子-内含子连接,诊断时或回顾性诊断样本的VAF截止率为1%。最小平均基本覆盖深度约为5000x,变体调用的最小覆盖阈值为1000x,至少需要10次变体读取。变异的致病性评估按照慢性淋巴细胞白血病(CLL)的ERIC指南和TP53变异报告[5]的推荐指南进行。从最新版本的UMD_TP53数据库(https://p53.fr/tp53-database)中的TP53单核苷酸多态性(SNP)数据中鉴定出多态性并排除(图S1)。对124名患者的外周血循环肿瘤细胞样本、11名患者的组织活检样本和5名患者的骨髓或其他液体样本进行测序。收集诊断时的临床和生物学特征、治疗细节和结果(表S1)。34%的患者(47/140)共检测到53种突变,其中6例患者有2种突变;由于该技术的高灵敏度(包括VAF截止率为1%的整个编码序列)和所分析样本的类型,该频率高于通常报道的频率,在大多数情况下,外周血单个核细胞(PBMC)具有高肿瘤细胞浸润。vaf明显高,64%的患者超过40%。10个突变的VAF小于10%,Sanger测序无法检测到。大多数突变为错义突变(74%),主要分布在DNA结合域,热点突变位于R278密码子(9/53突变)。 对68例患者进行了荧光原位杂交(FISH)分析,发现20例患者中有17p缺失,其中一半与TP53突变相关(图1A-C;表S1和表S2)。图1 TP53突变的临床影响。(A) TP53蛋白中TP53突变的分布。(B) TP53突变类型的比例,以占检测到的突变总数的百分比表示(n = 53)。(C)仅突变、仅17p缺失、突变和缺失均存在、无TP53异常的患者频率。频率计算相对于突变和缺失筛查病例总数(n = 68)。(D) Kaplan-Meier估计的总生存期(OS)基于是否存在TP53突变和无效或错义突变。(E) Kaplan-Meier根据TP53突变的存在(mut)或不存在(WT)以及HD阿糖胞苷或其他治疗的使用(tt)估计PFS。(F)与无异常患者相比,Kaplan-Meier基于是否存在TP53缺失和突变来估计OS。P值通过log-rank检验。在中位6.4年的随访中,140名患者中有76人死亡。tp53突变患者的总生存期(OS)低于未突变患者(中位OS: 2.45 vs. 9年)(p = 0.001)。我们接下来评估突变类型如何影响患者预后。错义突变导致异常p53蛋白的表达,并具有显性的负面影响,而零突变(无义突变、剪接位点突变和移码突变)导致p53完全不表达。TP53零突变和错义突变对生存的影响相同(图1D)。共有131例患者需要治疗,其中包括高剂量阿糖胞苷(RCHOP/RDHAP或RDHAX)作为主要治疗方式,然后在一部分患者中进行ASCT,这些患者均匀分布在tp53突变组和未突变组之间。在其余未接受任何治疗的9名患者中,只有2名携带TP53突变。一线治疗后,TP53突变患者的无进展生存期(PFS)也短于无突变患者(中位PFS: 0.58 vs 4.58年;p = 0.013)(表S1和图S2C), 1年PFS率为45%对80%。接受高剂量阿糖胞苷方案(p = 0.001)和其他治疗(r -苯达莫司汀、RCHOP或伊鲁替尼)的tp53突变患者的中位PFS比未突变患者短(p = 0.016)(图1E)。这些结果与临床试验结果一致;然而,TP53突变组在这些试验中代表性不足。与先前的报道一致,囊胚形态、Ki-67指数>; 30%和MIPI的高风险与较差的结果显著相关(p < 0.001, 0.002和<; 0.001)。值得注意的是,囊胚形态和Ki-67指数>; 30%都与TP53突变显著相关(p = 0.027和0.006,卡方检验)(图S2A)。根据MIPI的高风险患者在两组中分布均匀,因为患者年龄较大,主要表现为白血病(淋巴细胞计数>; 5 × 109/L和高白细胞计数),导致对进展风险的高估。MCL具有异质性细胞起源;大多数病例发生在生发前,特征是很少或没有IGHV体细胞突变,而其他病例发生在生发后中心,并与较高的体细胞IGHV突变负担相关。Navarro等人发表了突变的IGHV和非淋巴结性白血病的临床表现可能对应于一种行为更懒惰的疾病亚型。我们的研究重点是白血病MCL。首先分析IGHV突变状态,以确认克隆检测,并记录这些白血病患者的IGHV突变状态和IGHV基因使用的限制。通过NGS (LymphoTrack IGHV Leader体细胞超突变for MiSeq Illumina)分析IGHV基因重排,并通过Vidjil web应用程序(https://app.vidjil.org/)鉴定克隆型,以记录这些白血病病例并确定可能的惰性谱。突变的IGHV患者占患者的三分之一,在突变和未突变的TP53组中平均分配。IGHV重排谱偏斜,对IGHV基因的使用有很强的限制。最常见的重排是IGHV4-34(14%)、VH3-21(9%)、VH5-51(6%)和VH1-8(7%),这些重排以未突变为主。相反,VH3-33和VH4-39重排被发现含有突变。IGHV突变状态,无论是否使用98%或97%的身份阈值,也无论TP53状态如何,都不会影响生存或进展(图S2A,D)。这一发现与Yi等人的研究结果形成对比。 [6],这表明突变的IGHV基因在C1组中富集,并且与更有利的结果相关。此外,IGHV基因的使用对生存没有影响(图S2B)。尽管关于17p缺失存在的数据是不完整的,但他们能够识别出两组存活率不同的人。突变的存在,无论是单独存在还是与17p缺失联合存在(占所有患者的31%)(图1C),对生存也有类似的负面影响(图1F)。相比之下,单独缺失(占所有患者的15%)并没有显著影响生存,正如OS曲线所示,这与没有TP53异常的组(占所有患者的54%)没有显著差异。只有一名懒惰行为的患者有TP53无义突变和17p缺失。这些发现与先前的研究结果一致,并强调NGS是评估TP53异常以识别高危患者的强制性方法。总之,按照NCCN指南[7]的建议,所有入组临床试验的患者应在治疗前使用NGS进行TP53测序。这种方法对于确定复发/难治性疾病的危险群体和准确解释治疗效果结果至关重要。在我们的真实队列中,只有一半的患者在诊断时接受了TP53测序,而整个队列中有34%的患者表现出突变。与CLL不同,TP53突变状态尚未整合到MCL的治疗算法中。然而,随着这些数据的广泛获取,它们将使临床试验中根据TP53突变对患者进行分层,以评估在一线治疗中添加BTKi的价值。
本文章由计算机程序翻译,如有差异,请以英文原文为准。



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
