{"title":"A Rare Case of <i>PRKACA</i> Duplication-Associated Childhood-Onset Primary Pigmented Nodular Adrenocortical Disease.","authors":"Padala Ravi Kumar, Bandana Dash, Deepak Kumar Dash, Debasish Patro, Jatin Kumar Majhi, Bhabani Sankar Dhal","doi":"10.1210/jcemcr/luaf035","DOIUrl":null,"url":null,"abstract":"<p><p>Primary pigmented nodular adrenocortical disease (PPNAD) is a rare but important cause of adrenocorticotropic hormone (ACTH)-independent Cushing syndrome (CS). It usually presents as cyclical CS in young adults. Childhood onset of PPNAD is exceedingly rare. About 90% of cases of PPNAD are associated with Carney complex (CNC). Both PPNAD and CNC are linked to diverse pathogenic variants of the <i>PRKAR1A</i> gene, which encodes the regulatory subunit type 1 alpha of protein kinase A (PKA). Pathogenic variants of <i>PRKACA</i> gene, which encodes the catalytic subunit alpha of PKA, are extremely rare in PPNAD. We report a case of a female child, aged 8 years and 3 months, who presented with features suggestive of CS, including obesity, short stature, hypertension, moon facies, acne, and facial plethora but without classical striae or signs of CNC. Hormonal evaluation confirmed ACTH-independent CS. However, abdominal imaging revealed normal adrenal morphology. Genetic analysis identified a duplication of the <i>PRKACA</i> gene on chromosome 19p, which is linked to PPNAD. The patient underwent bilateral laparoscopic adrenalectomy, and histopathological study confirmed the PPNAD diagnosis. Postoperative follow-up showed resolution of cushingoid features and hypertension. To our knowledge, this is the first reported case of a female child with <i>PRKACA</i> duplication presenting as CS due to PPNAD.</p>","PeriodicalId":73540,"journal":{"name":"JCEM case reports","volume":"3 3","pages":"luaf035"},"PeriodicalIF":0.0000,"publicationDate":"2025-03-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11891440/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JCEM case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jcemcr/luaf035","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
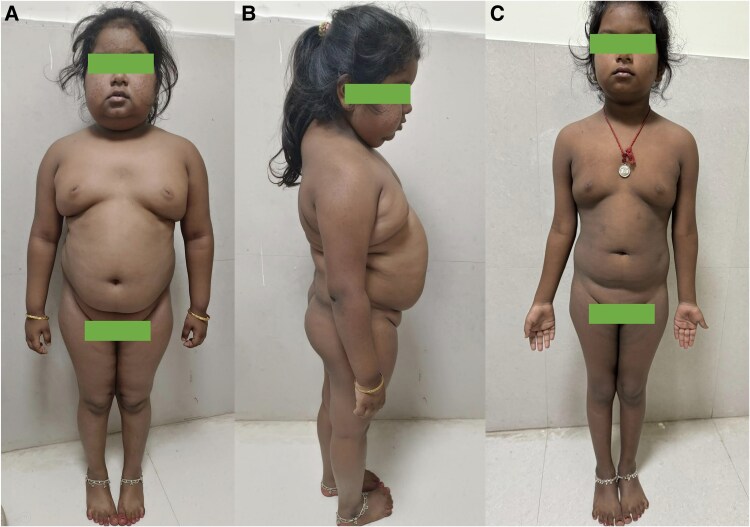
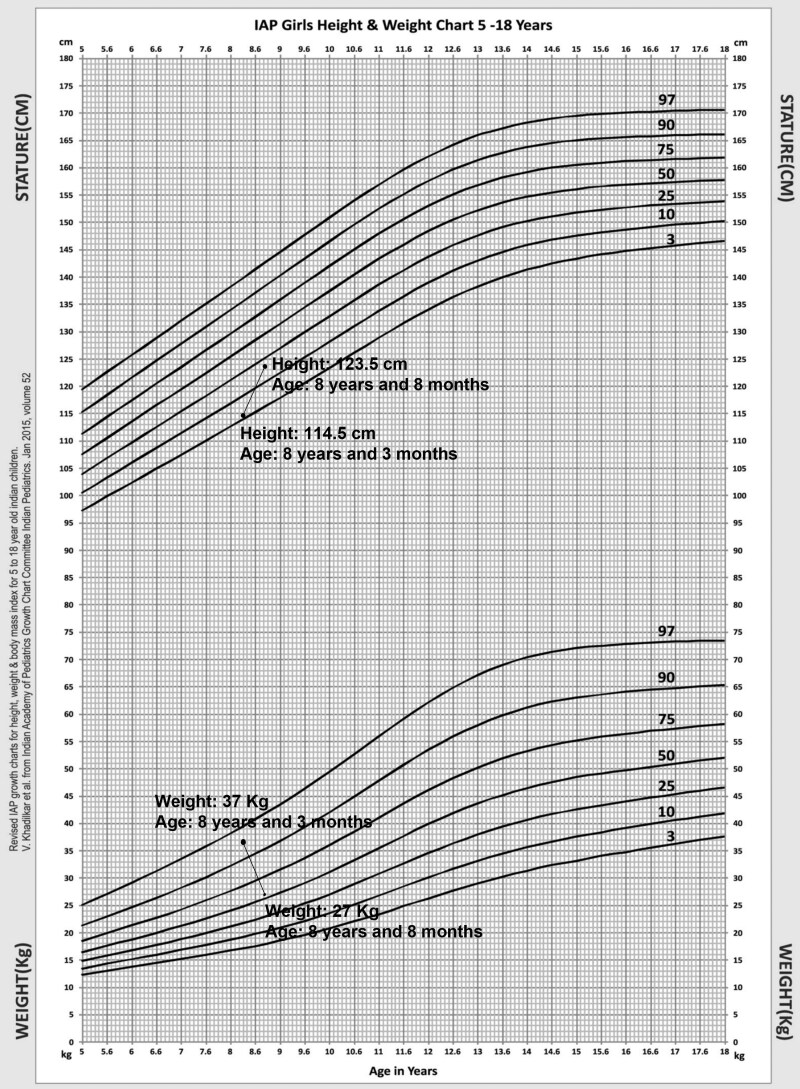
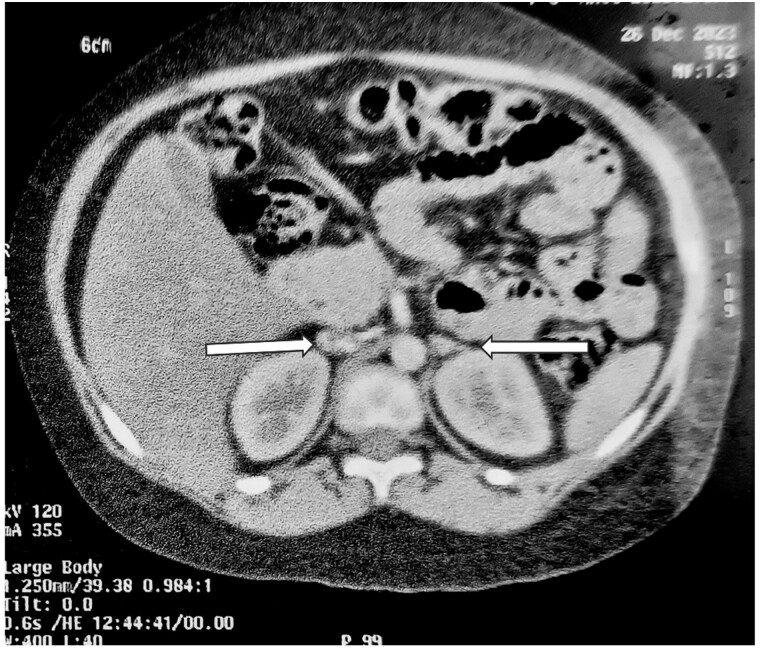
Primary pigmented nodular adrenocortical disease (PPNAD) is a rare but important cause of adrenocorticotropic hormone (ACTH)-independent Cushing syndrome (CS). It usually presents as cyclical CS in young adults. Childhood onset of PPNAD is exceedingly rare. About 90% of cases of PPNAD are associated with Carney complex (CNC). Both PPNAD and CNC are linked to diverse pathogenic variants of the PRKAR1A gene, which encodes the regulatory subunit type 1 alpha of protein kinase A (PKA). Pathogenic variants of PRKACA gene, which encodes the catalytic subunit alpha of PKA, are extremely rare in PPNAD. We report a case of a female child, aged 8 years and 3 months, who presented with features suggestive of CS, including obesity, short stature, hypertension, moon facies, acne, and facial plethora but without classical striae or signs of CNC. Hormonal evaluation confirmed ACTH-independent CS. However, abdominal imaging revealed normal adrenal morphology. Genetic analysis identified a duplication of the PRKACA gene on chromosome 19p, which is linked to PPNAD. The patient underwent bilateral laparoscopic adrenalectomy, and histopathological study confirmed the PPNAD diagnosis. Postoperative follow-up showed resolution of cushingoid features and hypertension. To our knowledge, this is the first reported case of a female child with PRKACA duplication presenting as CS due to PPNAD.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
