Research-Based Whole Genome Sequencing Identifies Biallelic Loss of Function Variants in DOCK3 Gene Causing DOCK3-Related Disorder: The End of a Diagnostic Journey for This Family
{"title":"Research-Based Whole Genome Sequencing Identifies Biallelic Loss of Function Variants in DOCK3 Gene Causing DOCK3-Related Disorder: The End of a Diagnostic Journey for This Family","authors":"Khurram Liaqat, Kayla Treat, Lili Mantcheva, Aaron McLaughlin, Amy Breman, Molly McPheron, Erin Conboy, Francesco Vetrini","doi":"10.1111/cge.14741","DOIUrl":null,"url":null,"abstract":"<p>The <i>DOCK3</i> gene (NM_004947.5) is located on chromosome 3p21.2 spanning 53 exons and encodes the dedicator of cytokinesis 3 protein. DOCK3 belongs to the family of guanine nucleotide exchange factors (GEFs) that activate GTPases. <i>DOCK3</i> is expressed almost exclusively in the central nervous system and has been shown to promote axonal outgrowth. Biallelic disruptions of <i>DOCK3</i> are implicated in a neurodevelopmental disorder presenting with intellectual disability, hypotonia and ataxia (OMIM: 618292). We report a 9-year-old female with global developmental delay, moderate intellectual disability, wide-based and ataxic gait, hypotonia, benign nocturnal myoclonus, bifid uvula, moderate obstructive sleep apnea, and alternating esotropia. Prior to enrollment in the Undiagnosed Rare Disease Clinic (URDC), the patient's clinical exome testing was negative. The subsequent enrollment in URDC allowed further research investigations through whole genome sequencing (GS) that identified two compound heterozygous variants in the <i>DOCK3</i> gene, ultimately yielding an unequivocal definitive molecular diagnosis.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"108 1","pages":"109-111"},"PeriodicalIF":2.3000,"publicationDate":"2025-03-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14741","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14741","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
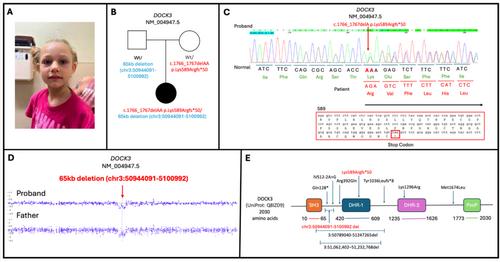
The DOCK3 gene (NM_004947.5) is located on chromosome 3p21.2 spanning 53 exons and encodes the dedicator of cytokinesis 3 protein. DOCK3 belongs to the family of guanine nucleotide exchange factors (GEFs) that activate GTPases. DOCK3 is expressed almost exclusively in the central nervous system and has been shown to promote axonal outgrowth. Biallelic disruptions of DOCK3 are implicated in a neurodevelopmental disorder presenting with intellectual disability, hypotonia and ataxia (OMIM: 618292). We report a 9-year-old female with global developmental delay, moderate intellectual disability, wide-based and ataxic gait, hypotonia, benign nocturnal myoclonus, bifid uvula, moderate obstructive sleep apnea, and alternating esotropia. Prior to enrollment in the Undiagnosed Rare Disease Clinic (URDC), the patient's clinical exome testing was negative. The subsequent enrollment in URDC allowed further research investigations through whole genome sequencing (GS) that identified two compound heterozygous variants in the DOCK3 gene, ultimately yielding an unequivocal definitive molecular diagnosis.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
