Ali Keshavarz Mohammadian, Negar Ashari Astani and Farzaneh Shayeganfar
{"title":"Computational design of dopant-free hole transporting materials: achieving an optimal balance between water stability and charge transport†","authors":"Ali Keshavarz Mohammadian, Negar Ashari Astani and Farzaneh Shayeganfar","doi":"10.1039/D5CP00082C","DOIUrl":null,"url":null,"abstract":"<p >Hole transporting materials (HTMs) play a crucial role in the performance and stability of perovskite solar cells (PSCs). The interaction of HTMs with water significantly affects the overall stability and efficiency of these devices. Hydrophilic HTMs or those lacking adequate water resistance can absorb moisture, leading to degradation of both the HTM and the perovskite layer. In this study, we employed a proof-of-principle approach to investigate the effect of various chemical modifications on a promising HTM candidate, 8,11-bis(4-(<em>N</em>,<em>N</em>-bis(4-methoxyphenyl)amino)-1-phenyl)-dithieno[1,2-<em>b</em>:4,3-<em>b</em>]phenazine (<strong>TQ4</strong>). Using molecular dynamics simulations, we examined the collective behavior of chemically modified <strong>TQ4</strong> molecules in the presence of water at different concentrations. To ensure that enhanced water resistance did not compromise the desirable electronic properties of the HTM, we analyzed both the individual and collective electronic structures of the HTM molecule and its molecular crystal. Additionally, we calculated the charge transport rate in different directions within the HTM crystal using Marcus theory. Our findings indicate that chemical modifications at the periphery of <strong>TQ4</strong>, particularly the symmetric addition of two F-chains, result in the optimal combination of electronic, crystal structure, and water-resistant properties. HOMO shape analysis reveals that the HOMO does not extend onto the added F-chains, reducing the maximum predicted hole mobility relative to <strong>TQ4</strong> by an order of magnitude. Despite this, a hole mobility of 2.8 × 10<small><sup>−4</sup></small> cm<small><sup>2</sup></small> V<small><sup>−1</sup></small> s<small><sup>−1</sup></small> is successfully achieved for all designed HTMs, reflecting a compromise between stability and charge transport. This atomistic insight into the collective behavior of chemically modified HTMs and its effect on hole transport pathways paves the way for designing more effective HTMs for PSC applications.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 18","pages":" 9511-9521"},"PeriodicalIF":2.9000,"publicationDate":"2025-04-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00082c","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
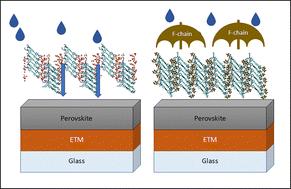
Hole transporting materials (HTMs) play a crucial role in the performance and stability of perovskite solar cells (PSCs). The interaction of HTMs with water significantly affects the overall stability and efficiency of these devices. Hydrophilic HTMs or those lacking adequate water resistance can absorb moisture, leading to degradation of both the HTM and the perovskite layer. In this study, we employed a proof-of-principle approach to investigate the effect of various chemical modifications on a promising HTM candidate, 8,11-bis(4-(N,N-bis(4-methoxyphenyl)amino)-1-phenyl)-dithieno[1,2-b:4,3-b]phenazine (TQ4). Using molecular dynamics simulations, we examined the collective behavior of chemically modified TQ4 molecules in the presence of water at different concentrations. To ensure that enhanced water resistance did not compromise the desirable electronic properties of the HTM, we analyzed both the individual and collective electronic structures of the HTM molecule and its molecular crystal. Additionally, we calculated the charge transport rate in different directions within the HTM crystal using Marcus theory. Our findings indicate that chemical modifications at the periphery of TQ4, particularly the symmetric addition of two F-chains, result in the optimal combination of electronic, crystal structure, and water-resistant properties. HOMO shape analysis reveals that the HOMO does not extend onto the added F-chains, reducing the maximum predicted hole mobility relative to TQ4 by an order of magnitude. Despite this, a hole mobility of 2.8 × 10−4 cm2 V−1 s−1 is successfully achieved for all designed HTMs, reflecting a compromise between stability and charge transport. This atomistic insight into the collective behavior of chemically modified HTMs and its effect on hole transport pathways paves the way for designing more effective HTMs for PSC applications.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
