Haozheng Li, Hanna H. Cramer, Jose B. Roque, Carlota Odena, Alex M. Shimozono, Paul J. Chirik
{"title":"The Role of Boron Reagents in Determining the Site-Selectivity of Pyridine(dicarbene) Cobalt-Catalyzed C–H Borylation of Fluorinated Arenes","authors":"Haozheng Li, Hanna H. Cramer, Jose B. Roque, Carlota Odena, Alex M. Shimozono, Paul J. Chirik","doi":"10.1021/jacs.4c15596","DOIUrl":null,"url":null,"abstract":"The origin of the <i>meta</i>- and <i>ortho</i>-to-fluorine site-selectivity in the C(sp<sup>2</sup>)–H borylation of fluorinated arenes with B<sub>2</sub>Pin<sub>2</sub> and HBPin promoted by pyridine(dicarbene)cobalt catalysts has been investigated. <i>In situ</i> generation of the cobalt(I)-boryl complex and treatment with three representative fluoroarenes established <i>meta</i>-selective C(sp<sup>2</sup>)–H oxidative addition to form predominantly the <i>meta</i> isomers of the corresponding cobalt(I)-aryl complexes. Attempts to observe or isolate the four-coordinate cobalt(I)-boryl complex yielded the cobalt-hydride dimer, <b>[(</b><sup><b>iPr</b></sup><b>ACNC)CoH]</b><sub><b>2</b></sub>, borohydride <b>(</b><sup><b>iPr</b></sup><b>ACNC)CoH</b><sub><b>2</b></sub><b>BPin</b>, or diboryl hydride, <b>(</b><sup><b>iPr</b></sup><b>ACNC)CoH(BPin)</b><sub><b>2</b></sub> depending on the amounts of B<sub>2</sub>Pin<sub>2</sub> and HBPin present. The phosphite derivatives <b>(</b><sup><b>iPr</b></sup><b>ACNC)CoH(P(O</b><sup><b>i</b></sup><b>Pr)</b><sub><b>3</b></sub><b>)</b> and <b>(</b><sup><b>iPr</b></sup><b>ACNC)CoBPin(P(O</b><sup><b>i</b></sup><b>Pr)</b><sub><b>3</b></sub><b>)</b> were prepared and crystallographically characterized. In the catalytic borylation of 1,3-difluorobenzene, <i>ortho</i>-to-fluorine cobalt(I)-aryl and borohydride complexes were identified as resting states despite <i>meta</i>-to-fluorine borylation being the major product of catalysis. Deuterium kinetic isotope effects support irreversible but not turnover-limiting C(sp<sup>2</sup>)–H oxidative addition. Stoichiometric borylation of isolated cobalt(I)-aryl intermediates with B<sub>2</sub>Pin<sub>2</sub> established that the <i>meta-</i>cobalt(I)-aryl was more reactive than the <i>ortho-</i>isomer and accounts for the observed cobalt(I)-aryl resting states. All cobalt(I)-aryl compounds reacted more quickly with HBPin. While <i>ortho</i>-cobalt(I)-aryl compounds yielded arylboronate products with high site-selectivity, <i>meta</i>-cobalt-aryl counterparts yielded a mixture of arylboronate isomers and free arene. Deuterium labeling experiments with DBPin confirmed that HBPin mediates reversible C(sp<sup>2</sup>)–H oxidative addition. Thus, the overall site-selectivity arises from two reinforcing effects: (<i>i</i>) kinetically <i>meta-</i>selective oxidative addition and (<i>ii</i>) faster reaction of the <i>meta</i>-cobalt-aryl isomer with B<sub>2</sub>Pin<sub>2</sub>. As B<sub>2</sub>Pin<sub>2</sub> is converted to HBPin, C(sp<sup>2</sup>)–H reductive elimination competes against borylation of the <i>meta</i>-cobalt-aryl isomer, resulting in increased <i>ortho</i>-selective borylation.","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"29 1","pages":""},"PeriodicalIF":15.6000,"publicationDate":"2025-04-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/jacs.4c15596","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
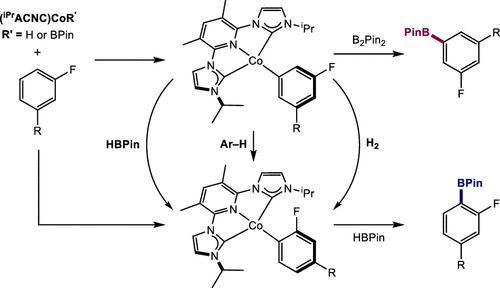
The origin of the meta- and ortho-to-fluorine site-selectivity in the C(sp2)–H borylation of fluorinated arenes with B2Pin2 and HBPin promoted by pyridine(dicarbene)cobalt catalysts has been investigated. In situ generation of the cobalt(I)-boryl complex and treatment with three representative fluoroarenes established meta-selective C(sp2)–H oxidative addition to form predominantly the meta isomers of the corresponding cobalt(I)-aryl complexes. Attempts to observe or isolate the four-coordinate cobalt(I)-boryl complex yielded the cobalt-hydride dimer, [(iPrACNC)CoH]2, borohydride (iPrACNC)CoH2BPin, or diboryl hydride, (iPrACNC)CoH(BPin)2 depending on the amounts of B2Pin2 and HBPin present. The phosphite derivatives (iPrACNC)CoH(P(OiPr)3) and (iPrACNC)CoBPin(P(OiPr)3) were prepared and crystallographically characterized. In the catalytic borylation of 1,3-difluorobenzene, ortho-to-fluorine cobalt(I)-aryl and borohydride complexes were identified as resting states despite meta-to-fluorine borylation being the major product of catalysis. Deuterium kinetic isotope effects support irreversible but not turnover-limiting C(sp2)–H oxidative addition. Stoichiometric borylation of isolated cobalt(I)-aryl intermediates with B2Pin2 established that the meta-cobalt(I)-aryl was more reactive than the ortho-isomer and accounts for the observed cobalt(I)-aryl resting states. All cobalt(I)-aryl compounds reacted more quickly with HBPin. While ortho-cobalt(I)-aryl compounds yielded arylboronate products with high site-selectivity, meta-cobalt-aryl counterparts yielded a mixture of arylboronate isomers and free arene. Deuterium labeling experiments with DBPin confirmed that HBPin mediates reversible C(sp2)–H oxidative addition. Thus, the overall site-selectivity arises from two reinforcing effects: (i) kinetically meta-selective oxidative addition and (ii) faster reaction of the meta-cobalt-aryl isomer with B2Pin2. As B2Pin2 is converted to HBPin, C(sp2)–H reductive elimination competes against borylation of the meta-cobalt-aryl isomer, resulting in increased ortho-selective borylation.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
