{"title":"Thermodynamic Favorability of the 1T Phase over the 1H Phase in Group III Metal Monochalcogenide Zigzag Nanoribbons","authors":"Emin Aliyev, Arash Mobaraki, Hâldun Sevinçli, Seymur Jahangirov","doi":"10.1021/acs.jpcc.5c00765","DOIUrl":null,"url":null,"abstract":"Owing to the promising optoelectronic and thermoelectric properties of two-dimensional (2D) group III–VI materials (MXs), their nanoribbons (NRs) have attracted notable attention as an emerging class of quasi-one-dimensional (quasi-1D) nanostructures. Due to the fact that the most stable 2D monolayer polymorph of MXs is the 1H phase, to date, existing studies in the literature have predominantly focused on the NRs formed from 1H phase MXs. Nevertheless, NRs of the 1T phase have received little to no attention. Employing ab initio simulations based on density functional theory, we systematically compared the thermodynamic stability of hydrogen-passivated and unpassivated 1T and 1H ZNRs of GaS, GaSe, and InSe. Our results reveal that nonpolar 1T phase MX ZNRs are thermodynamically more favorable than polar 1H MX ZNRs at widths up to 34 nm, a range that is realizable through contemporary experimental fabrication techniques. Furthermore, unlike metallic 1H ZNRs, 1T ZNRs remain semiconductors and retain Mexican-hat-shaped top valence bands. Complementarily, hydrogenation energies of 1T InSe ZNRs are positive, and due to the edge-localized states, the 1T unpassivated ZNRs possess nearly flat top valence bands. Our findings serve as a compass for subsequent synthesis pathways of group III–VI NRs.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"12 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2025-04-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.5c00765","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
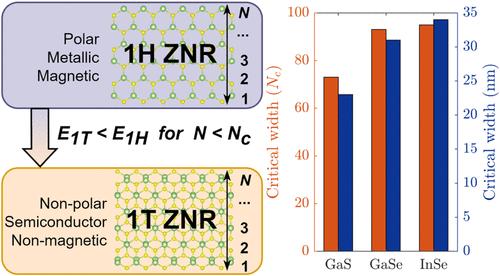
Owing to the promising optoelectronic and thermoelectric properties of two-dimensional (2D) group III–VI materials (MXs), their nanoribbons (NRs) have attracted notable attention as an emerging class of quasi-one-dimensional (quasi-1D) nanostructures. Due to the fact that the most stable 2D monolayer polymorph of MXs is the 1H phase, to date, existing studies in the literature have predominantly focused on the NRs formed from 1H phase MXs. Nevertheless, NRs of the 1T phase have received little to no attention. Employing ab initio simulations based on density functional theory, we systematically compared the thermodynamic stability of hydrogen-passivated and unpassivated 1T and 1H ZNRs of GaS, GaSe, and InSe. Our results reveal that nonpolar 1T phase MX ZNRs are thermodynamically more favorable than polar 1H MX ZNRs at widths up to 34 nm, a range that is realizable through contemporary experimental fabrication techniques. Furthermore, unlike metallic 1H ZNRs, 1T ZNRs remain semiconductors and retain Mexican-hat-shaped top valence bands. Complementarily, hydrogenation energies of 1T InSe ZNRs are positive, and due to the edge-localized states, the 1T unpassivated ZNRs possess nearly flat top valence bands. Our findings serve as a compass for subsequent synthesis pathways of group III–VI NRs.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
