Eran Dvir, Anat Elman, Danielle Simmons, Israel Shapiro, Revital Duvdevani, Arik Dahan, Amnon Hoffman, Jonathan E. Friedman
{"title":"DP-155, a Lecithin Derivative of Indomethacin, is a Novel Nonsteroidal Antiinflammatory Drug for Analgesia and Alzheimer's Disease Therapy","authors":"Eran Dvir, Anat Elman, Danielle Simmons, Israel Shapiro, Revital Duvdevani, Arik Dahan, Amnon Hoffman, Jonathan E. Friedman","doi":"10.1111/j.1527-3458.2007.00014.x","DOIUrl":null,"url":null,"abstract":"<p>DP-155 is a lipid prodrug of indomethacin that comprises the latter conjugated to lecithin at position <i>sn</i>-2 via a 5-carbon length linker. It is cleaved by phospholipase A<sub>2</sub> (PLA)<sub>2</sub> to a greater extent than similar compounds with linkers of 2, 3, and 4 carbons. Indomethacin is the principal metabolite of DP-155 in rat serum and, after DP-155 oral administration, the half-life of the metabolite was 22 and 93 h in serum and brain, respectively, compared to 10 and 24 h following indomethacin administration. The brain to serum ratio was 3.5 times higher for DP-155 than for indomethacin. <i>In vitro</i> studies demonstrated that DP-155 is a selective cyclooxygenase (COX)-2 inhibitor. After it is cleaved, its indomethacin derivative nonselectively inhibits both COX-1 and -2. DP-155 showed a better toxicity profile probably due to the sustained, low serum levels and reduced maximal concentration of its indomethacin metabolite. DP-155 did not produce gastric toxicity at the highest acute dose tested (0.28 mmol/kg), while indomethacin caused gastric ulcers at a dose 33-fold lower. Furthermore, after repeated oral dosing, gastrointestinal and renal toxicity was lower (10- and 5-fold, respectively) and delayed with DP-155 compared to indomethacin. In addition to reduced toxicity, DP-155 had similar ameliorative effects to indomethacin in antipyretic and analgesia models. Moreover, DP-155 and indomethacin were equally efficacious in reducing levels of amyloid ß (Aß)42 in transgenic Alzheimer's disease mouse (Tg2576) brains as well as reducing Aß42 intracellular uptake, neurodegeneration, and inflammation in an <i>in vitro</i> AD model. The relatively high brain levels of indomethacin after DP-155 administration explain the equal efficacy of DP-155 despite its low systemic blood concentrations. Compared to indomethacin, the favored safety profile and equal efficacy of DP-155 establish the compound as a potential candidate for chronic use to treat AD-related pathology and for analgesia.</p>","PeriodicalId":94307,"journal":{"name":"CNS drug reviews","volume":"13 2","pages":"260-277"},"PeriodicalIF":0.0000,"publicationDate":"2007-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1111/j.1527-3458.2007.00014.x","citationCount":"29","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"CNS drug reviews","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/j.1527-3458.2007.00014.x","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 29
Abstract
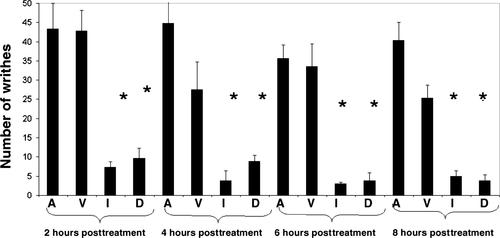
DP-155 is a lipid prodrug of indomethacin that comprises the latter conjugated to lecithin at position sn-2 via a 5-carbon length linker. It is cleaved by phospholipase A2 (PLA)2 to a greater extent than similar compounds with linkers of 2, 3, and 4 carbons. Indomethacin is the principal metabolite of DP-155 in rat serum and, after DP-155 oral administration, the half-life of the metabolite was 22 and 93 h in serum and brain, respectively, compared to 10 and 24 h following indomethacin administration. The brain to serum ratio was 3.5 times higher for DP-155 than for indomethacin. In vitro studies demonstrated that DP-155 is a selective cyclooxygenase (COX)-2 inhibitor. After it is cleaved, its indomethacin derivative nonselectively inhibits both COX-1 and -2. DP-155 showed a better toxicity profile probably due to the sustained, low serum levels and reduced maximal concentration of its indomethacin metabolite. DP-155 did not produce gastric toxicity at the highest acute dose tested (0.28 mmol/kg), while indomethacin caused gastric ulcers at a dose 33-fold lower. Furthermore, after repeated oral dosing, gastrointestinal and renal toxicity was lower (10- and 5-fold, respectively) and delayed with DP-155 compared to indomethacin. In addition to reduced toxicity, DP-155 had similar ameliorative effects to indomethacin in antipyretic and analgesia models. Moreover, DP-155 and indomethacin were equally efficacious in reducing levels of amyloid ß (Aß)42 in transgenic Alzheimer's disease mouse (Tg2576) brains as well as reducing Aß42 intracellular uptake, neurodegeneration, and inflammation in an in vitro AD model. The relatively high brain levels of indomethacin after DP-155 administration explain the equal efficacy of DP-155 despite its low systemic blood concentrations. Compared to indomethacin, the favored safety profile and equal efficacy of DP-155 establish the compound as a potential candidate for chronic use to treat AD-related pathology and for analgesia.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
