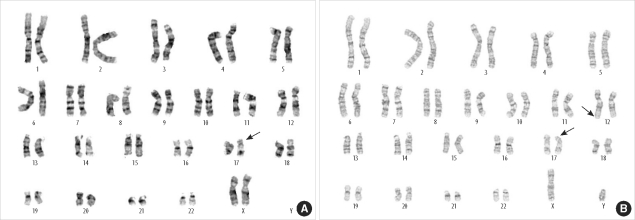
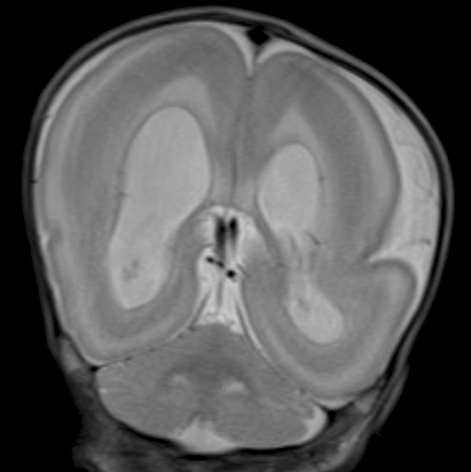
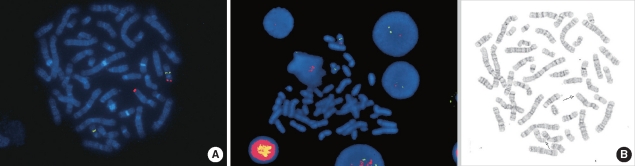
Miller-Dieker syndrome with der(17)t(12;17)(q24.33;p13.3)pat presenting with a potential risk of mis-identification as a de novo submicroscopic deletion of 17p13.3.
Young Jin Kim, Shin Yun Byun, Seon A Jo, Yong Beom Shin, Eun Hae Cho, Eun Yup Lee, Sang-Hyun Hwang
{"title":"Miller-Dieker syndrome with der(17)t(12;17)(q24.33;p13.3)pat presenting with a potential risk of mis-identification as a de novo submicroscopic deletion of 17p13.3.","authors":"Young Jin Kim, Shin Yun Byun, Seon A Jo, Yong Beom Shin, Eun Hae Cho, Eun Yup Lee, Sang-Hyun Hwang","doi":"10.3343/kjlm.2011.31.1.49","DOIUrl":null,"url":null,"abstract":"<p><p>Miller-Dieker syndrome involves a severe type of lissencephaly, which is caused by defects in the lissencephaly gene (LIS1). We report the case of a female infant with der(17)t(12;17)(q24.33;p13.3)pat caused by an unbalanced segregation of the parental balanced translocation of 17p with other chromosomes. The proband presented with facial dysmorphism, arthrogryposis, and intrauterine growth retardation. Most cases of Miller-Dieker syndrome have a de novo deletion involving 17p13.3. When Miller-Dieker syndrome is caused by an unbalanced translocation, mild-to-severe phenotypes occur according to the extension of the involved partner chromosome. However, a pure partial monosomy derived from a paternal balanced translocation is relatively rare. In this case, the submicroscopic cryptic deletion in the proband was initially elucidated by FISH, and karyotype analysis did not reveal additional chromosome abnormalities such as translocation. However, a family history of recurrent pregnancy abnormalities strongly suggested familial translocation. Sequential G-banding and FISH analysis of the father's chromosomes showed that the segment of 17p13.3→pter was attached to the 12qter. Thus, we report a case that showed resemblance to the findings in cases of a nearly pure 17p deletion, derived from t(12;17), and delineated by whole genome array comparative genomic hybridization (CGH). If such cases are incorrectly diagnosed as Miller-Dieker syndrome caused by de novo 17p13.3 deletion, the resultant improper genetic counseling may make it difficult to exactly predict the potential risk of recurrent lissencephaly for successive pregnancies.</p>","PeriodicalId":17890,"journal":{"name":"Korean Journal of Laboratory Medicine","volume":"31 1","pages":"49-53"},"PeriodicalIF":0.0000,"publicationDate":"2011-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3343/kjlm.2011.31.1.49","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Korean Journal of Laboratory Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3343/kjlm.2011.31.1.49","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 6
Abstract
Miller-Dieker syndrome involves a severe type of lissencephaly, which is caused by defects in the lissencephaly gene (LIS1). We report the case of a female infant with der(17)t(12;17)(q24.33;p13.3)pat caused by an unbalanced segregation of the parental balanced translocation of 17p with other chromosomes. The proband presented with facial dysmorphism, arthrogryposis, and intrauterine growth retardation. Most cases of Miller-Dieker syndrome have a de novo deletion involving 17p13.3. When Miller-Dieker syndrome is caused by an unbalanced translocation, mild-to-severe phenotypes occur according to the extension of the involved partner chromosome. However, a pure partial monosomy derived from a paternal balanced translocation is relatively rare. In this case, the submicroscopic cryptic deletion in the proband was initially elucidated by FISH, and karyotype analysis did not reveal additional chromosome abnormalities such as translocation. However, a family history of recurrent pregnancy abnormalities strongly suggested familial translocation. Sequential G-banding and FISH analysis of the father's chromosomes showed that the segment of 17p13.3→pter was attached to the 12qter. Thus, we report a case that showed resemblance to the findings in cases of a nearly pure 17p deletion, derived from t(12;17), and delineated by whole genome array comparative genomic hybridization (CGH). If such cases are incorrectly diagnosed as Miller-Dieker syndrome caused by de novo 17p13.3 deletion, the resultant improper genetic counseling may make it difficult to exactly predict the potential risk of recurrent lissencephaly for successive pregnancies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
