Vladimir Lazarevic, Katrine Whiteson, Nadia Gaïa, Yann Gizard, David Hernandez, Laurent Farinelli, Magne Osterås, Patrice François, Jacques Schrenzel
{"title":"Analysis of the salivary microbiome using culture-independent techniques.","authors":"Vladimir Lazarevic, Katrine Whiteson, Nadia Gaïa, Yann Gizard, David Hernandez, Laurent Farinelli, Magne Osterås, Patrice François, Jacques Schrenzel","doi":"10.1186/2043-9113-2-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The salivary microbiota is a potential diagnostic indicator of several diseases. Culture-independent techniques are required to study the salivary microbial community since many of its members have not been cultivated.</p><p><strong>Methods: </strong>We explored the bacterial community composition in the saliva sample using metagenomic whole genome shotgun (WGS) sequencing, the extraction of 16S rRNA gene fragments from metagenomic sequences (16S-WGS) and high-throughput sequencing of PCR-amplified bacterial 16S rDNA gene (16S-HTS) regions V1 and V3.</p><p><strong>Results: </strong>The hierarchical clustering of data based on the relative abundance of bacterial genera revealed that distances between 16S-HTS datasets for V1 and V3 regions were greater than those obtained for the same V region with different numbers of PCR cycles. Datasets generated by 16S-HTS and 16S-WGS were even more distant. Finally, comparison of WGS and 16S-based datasets revealed the highest dissimilarity.The analysis of the 16S-HTS, WGS and 16S-WGS datasets revealed 206, 56 and 39 bacterial genera, respectively, 124 of which have not been previously identified in salivary microbiomes. A large fraction of DNA extracted from saliva corresponded to human DNA. Based on sequence similarity search against completely sequenced genomes, bacterial and viral sequences represented 0.73% and 0.0036% of the salivary metagenome, respectively. Several sequence reads were identified as parts of the human herpesvirus 7.</p><p><strong>Conclusions: </strong>Analysis of the salivary metagenome may have implications in diagnostics e.g. in detection of microorganisms and viruses without designing specific tests for each pathogen.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":"2 ","pages":"4"},"PeriodicalIF":0.0000,"publicationDate":"2012-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2043-9113-2-4","citationCount":"62","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2043-9113-2-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 62
Abstract
Background: The salivary microbiota is a potential diagnostic indicator of several diseases. Culture-independent techniques are required to study the salivary microbial community since many of its members have not been cultivated.
Methods: We explored the bacterial community composition in the saliva sample using metagenomic whole genome shotgun (WGS) sequencing, the extraction of 16S rRNA gene fragments from metagenomic sequences (16S-WGS) and high-throughput sequencing of PCR-amplified bacterial 16S rDNA gene (16S-HTS) regions V1 and V3.
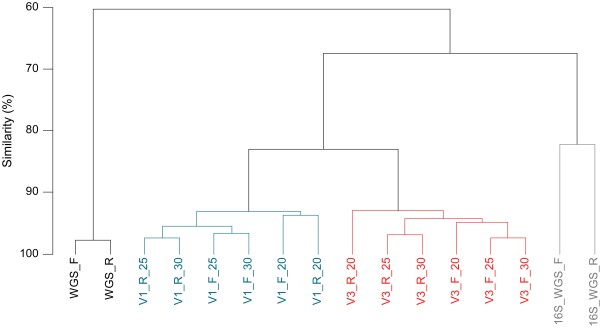
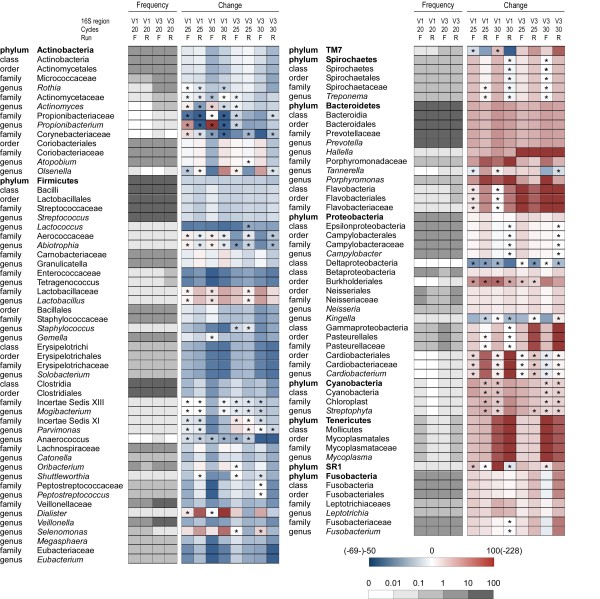
Results: The hierarchical clustering of data based on the relative abundance of bacterial genera revealed that distances between 16S-HTS datasets for V1 and V3 regions were greater than those obtained for the same V region with different numbers of PCR cycles. Datasets generated by 16S-HTS and 16S-WGS were even more distant. Finally, comparison of WGS and 16S-based datasets revealed the highest dissimilarity.The analysis of the 16S-HTS, WGS and 16S-WGS datasets revealed 206, 56 and 39 bacterial genera, respectively, 124 of which have not been previously identified in salivary microbiomes. A large fraction of DNA extracted from saliva corresponded to human DNA. Based on sequence similarity search against completely sequenced genomes, bacterial and viral sequences represented 0.73% and 0.0036% of the salivary metagenome, respectively. Several sequence reads were identified as parts of the human herpesvirus 7.
Conclusions: Analysis of the salivary metagenome may have implications in diagnostics e.g. in detection of microorganisms and viruses without designing specific tests for each pathogen.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
