{"title":"Base-By-Base version 2: single nucleotide-level analysis of whole viral genome alignments.","authors":"William Hillary, Song-Han Lin, Chris Upton","doi":"10.1186/2042-5783-1-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Base-By-Base is a Java-based multiple sequence alignment editor. It is capable of working with protein and DNA molecules, but many of its unique features relate to the manipulation of the genomes of large DNA viruses such as poxviruses, herpesviruses, baculoviruses and asfarviruses (1-400 kb). The tool was built to serve as a platform for comparative genomics at the level of individual nucleotides.</p><p><strong>Results: </strong>In version 2, BBB-v2, of Base-By-Base we have added a series of new features aimed at providing the bench virologist with a better platform to view, annotate and analyze these complex genomes. Although a poxvirus genome, for example, may be less than 200 kb, it probably encodes close to 200 proteins using multiple classes of promoters with frequent overlapping of promoters and coding sequences and even some overlapping of genes. The new features allow users to 1) add primer annotations or other data sets in batch mode, 2) export differences between sequences to other genome browsers, 3) compare multiple genomes at a single nucleotide level of detail, 4) create new alignments from subsets/subsequences of a very large master alignment and 5) allow display of summaries of deep RNA sequencing data sets on a genome sequence.</p><p><strong>Conclusion: </strong>BBB-v2 significantly improves the ability of virologists to work with genome sequences and provides a platform with which they can use a multiple sequence alignment as the basis for their own editable documents. Also, a .bbb document, with a variety of annotations in addition to the basic coding regions, can be shared among collaborators or made available to an entire research community. The program is available via Virology.ca using Java Web Start and is platform independent; the Java 1.5 virtual machine is required.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":"1 1","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2011-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-1-2","citationCount":"44","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-1-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 44
Abstract
Background: Base-By-Base is a Java-based multiple sequence alignment editor. It is capable of working with protein and DNA molecules, but many of its unique features relate to the manipulation of the genomes of large DNA viruses such as poxviruses, herpesviruses, baculoviruses and asfarviruses (1-400 kb). The tool was built to serve as a platform for comparative genomics at the level of individual nucleotides.
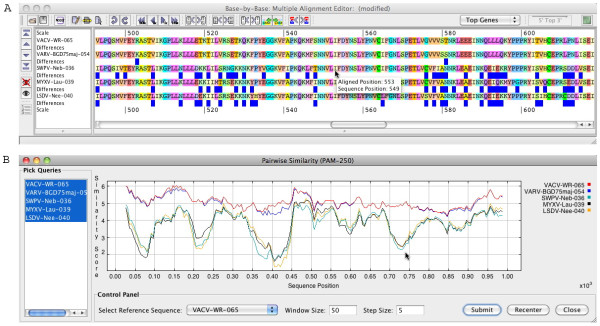
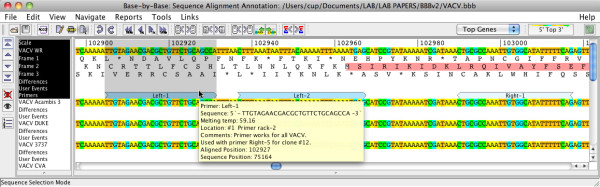
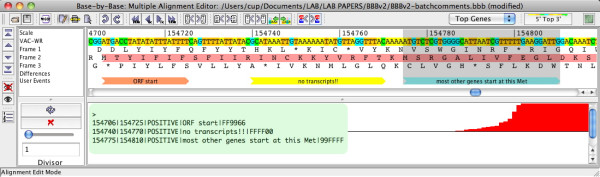
Results: In version 2, BBB-v2, of Base-By-Base we have added a series of new features aimed at providing the bench virologist with a better platform to view, annotate and analyze these complex genomes. Although a poxvirus genome, for example, may be less than 200 kb, it probably encodes close to 200 proteins using multiple classes of promoters with frequent overlapping of promoters and coding sequences and even some overlapping of genes. The new features allow users to 1) add primer annotations or other data sets in batch mode, 2) export differences between sequences to other genome browsers, 3) compare multiple genomes at a single nucleotide level of detail, 4) create new alignments from subsets/subsequences of a very large master alignment and 5) allow display of summaries of deep RNA sequencing data sets on a genome sequence.
Conclusion: BBB-v2 significantly improves the ability of virologists to work with genome sequences and provides a platform with which they can use a multiple sequence alignment as the basis for their own editable documents. Also, a .bbb document, with a variety of annotations in addition to the basic coding regions, can be shared among collaborators or made available to an entire research community. The program is available via Virology.ca using Java Web Start and is platform independent; the Java 1.5 virtual machine is required.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
