Ali Kord Valeshabad, Abdolmotaleb Mazidi, Reza Kord Valeshabad, Elham Imani, Hadi Kord, Mohammad Koohkan, Zrynal Sayinar, Khalil Al-Talib
{"title":"Papillon-lefèvre syndrome: a series of six cases in the same family.","authors":"Ali Kord Valeshabad, Abdolmotaleb Mazidi, Reza Kord Valeshabad, Elham Imani, Hadi Kord, Mohammad Koohkan, Zrynal Sayinar, Khalil Al-Talib","doi":"10.5402/2012/139104","DOIUrl":null,"url":null,"abstract":"<p><p>Papillon-Lefèvre syndrome (PLS) is a rare, autosomal recessive heterogeneous disorder, which is characterized by palmoplantar hyperkeratosis, early loss of primary and permanent teeth, and associated calcification of the dura mater. Herein we described six cases of PLS in the same family. In this series, six cases (two females and four males) with the mean age of 15.6 ± 10.4 years were recruited. Palmoplantar hyperkeratosis was detected in all of the cases, leading to a difficult and painful walking in two cases due to lesions on the soles. Skin lesions were sharply distinct from adjacent normal skin in all cases. Other skin lesions were located in the external malleolus (5/6), knee (4/6), elbow (4/6), toe and dorsal fingers (3/6), and the thighs (2/6). In three cases, all permanent teeth were exfoliated. In three others, no primary teeth remained. Severe gingivitis was observed in three patients. Radiologic study confirmed alveolar bone destruction in five cases. Delayed diagnosis and insufficient treatment of PLS patients can affect patient's life of by causing edentulism at a young age and may impose PLS patients to increased risk of social, psychological, and economical burdens.</p>","PeriodicalId":14682,"journal":{"name":"ISRN Dermatology","volume":"2012 ","pages":"139104"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.5402/2012/139104","citationCount":"10","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN Dermatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5402/2012/139104","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2012/12/3 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 10
Abstract
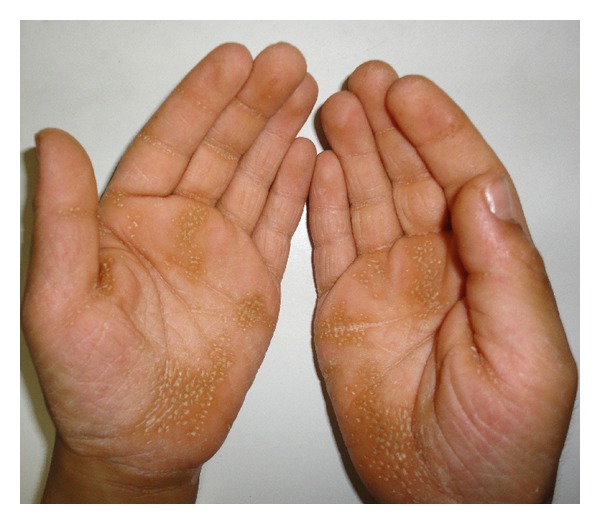
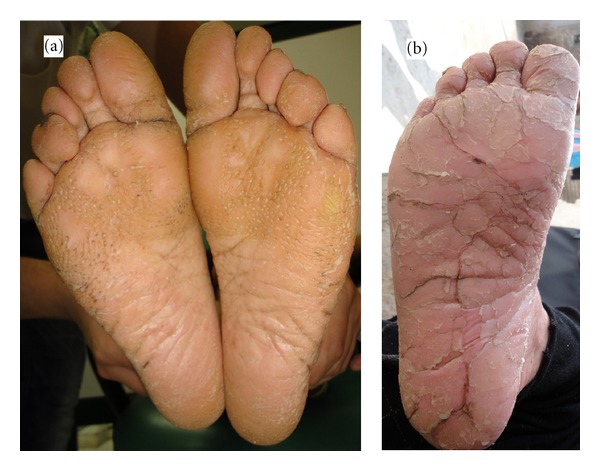
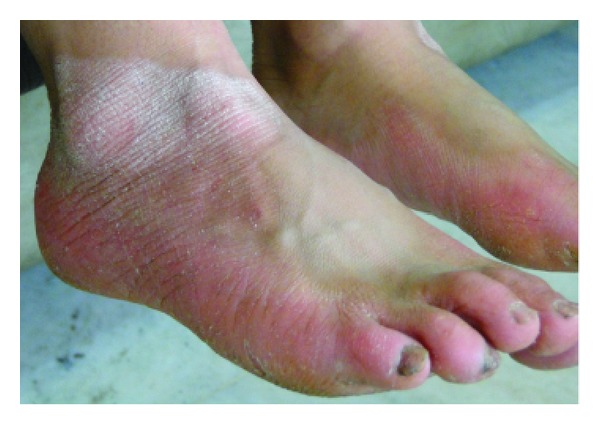
Papillon-Lefèvre syndrome (PLS) is a rare, autosomal recessive heterogeneous disorder, which is characterized by palmoplantar hyperkeratosis, early loss of primary and permanent teeth, and associated calcification of the dura mater. Herein we described six cases of PLS in the same family. In this series, six cases (two females and four males) with the mean age of 15.6 ± 10.4 years were recruited. Palmoplantar hyperkeratosis was detected in all of the cases, leading to a difficult and painful walking in two cases due to lesions on the soles. Skin lesions were sharply distinct from adjacent normal skin in all cases. Other skin lesions were located in the external malleolus (5/6), knee (4/6), elbow (4/6), toe and dorsal fingers (3/6), and the thighs (2/6). In three cases, all permanent teeth were exfoliated. In three others, no primary teeth remained. Severe gingivitis was observed in three patients. Radiologic study confirmed alveolar bone destruction in five cases. Delayed diagnosis and insufficient treatment of PLS patients can affect patient's life of by causing edentulism at a young age and may impose PLS patients to increased risk of social, psychological, and economical burdens.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
