Dario Di Silvestre, Italo Zoppis, Francesca Brambilla, Valeria Bellettato, Giancarlo Mauri, Pierluigi Mauri
{"title":"Availability of MudPIT data for classification of biological samples.","authors":"Dario Di Silvestre, Italo Zoppis, Francesca Brambilla, Valeria Bellettato, Giancarlo Mauri, Pierluigi Mauri","doi":"10.1186/2043-9113-3-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Unlabelled: </strong></p><p><strong>Background: </strong>Mass spectrometry is an important analytical tool for clinical proteomics. Primarily employed for biomarker discovery, it is increasingly used for developing methods which may help to provide unambiguous diagnosis of biological samples. In this context, we investigated the classification of phenotypes by applying support vector machine (SVM) on experimental data obtained by MudPIT approach. In particular, we compared the performance capabilities of SVM by using two independent collection of complex samples and different data-types, such as mass spectra (m/z), peptides and proteins.</p><p><strong>Results: </strong>Globally, protein and peptide data allowed a better discriminant informative content than experimental mass spectra (overall accuracy higher than 87% in both collection 1 and 2). These results indicate that sequencing of peptides and proteins reduces the experimental noise affecting the raw mass spectra, and allows the extraction of more informative features available for the effective classification of samples. In addition, proteins and peptides features selected by SVM matched for 80% with the differentially expressed proteins identified by the MAProMa software.</p><p><strong>Conclusions: </strong>These findings confirm the availability of the most label-free quantitative methods based on processing of spectral count and SEQUEST-based SCORE values. On the other hand, it stresses the usefulness of MudPIT data for a correct grouping of sample phenotypes, by applying both supervised and unsupervised learning algorithms. This capacity permit the evaluation of actual samples and it is a good starting point to translate proteomic methodology to clinical application.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":" ","pages":"1"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2043-9113-3-1","citationCount":"17","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2043-9113-3-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 17
Abstract
Unlabelled:
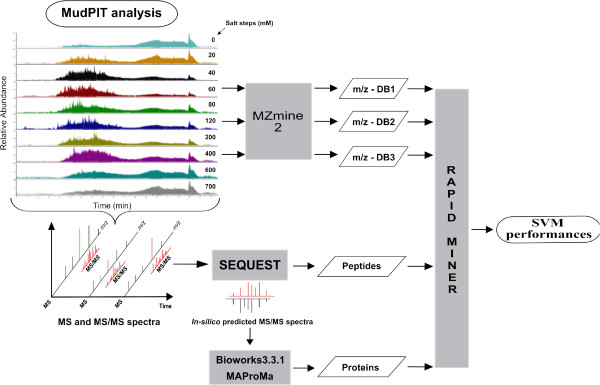
Background: Mass spectrometry is an important analytical tool for clinical proteomics. Primarily employed for biomarker discovery, it is increasingly used for developing methods which may help to provide unambiguous diagnosis of biological samples. In this context, we investigated the classification of phenotypes by applying support vector machine (SVM) on experimental data obtained by MudPIT approach. In particular, we compared the performance capabilities of SVM by using two independent collection of complex samples and different data-types, such as mass spectra (m/z), peptides and proteins.
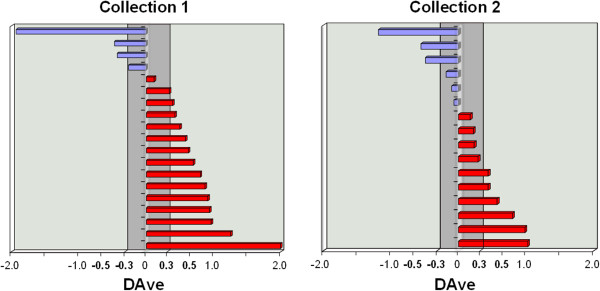
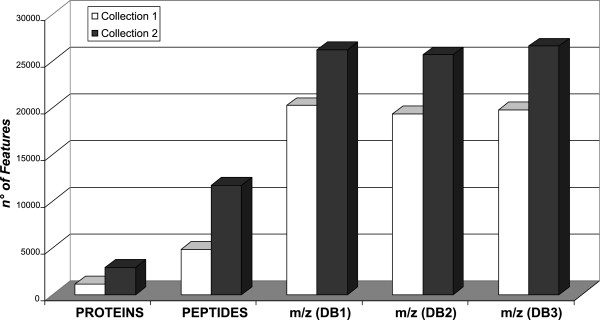
Results: Globally, protein and peptide data allowed a better discriminant informative content than experimental mass spectra (overall accuracy higher than 87% in both collection 1 and 2). These results indicate that sequencing of peptides and proteins reduces the experimental noise affecting the raw mass spectra, and allows the extraction of more informative features available for the effective classification of samples. In addition, proteins and peptides features selected by SVM matched for 80% with the differentially expressed proteins identified by the MAProMa software.
Conclusions: These findings confirm the availability of the most label-free quantitative methods based on processing of spectral count and SEQUEST-based SCORE values. On the other hand, it stresses the usefulness of MudPIT data for a correct grouping of sample phenotypes, by applying both supervised and unsupervised learning algorithms. This capacity permit the evaluation of actual samples and it is a good starting point to translate proteomic methodology to clinical application.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
