Richard E Higgs, Jon P Butler, Bomie Han, Michael D Knierman
{"title":"Quantitative Proteomics via High Resolution MS Quantification: Capabilities and Limitations.","authors":"Richard E Higgs, Jon P Butler, Bomie Han, Michael D Knierman","doi":"10.1155/2013/674282","DOIUrl":null,"url":null,"abstract":"<p><p>Recent improvements in the mass accuracy and resolution of mass spectrometers have led to renewed interest in label-free quantification using data from the primary mass spectrum (MS1) acquired from data-dependent proteomics experiments. The capacity for higher specificity quantification of peptides from samples enriched for proteins of biological interest offers distinct advantages for hypothesis generating experiments relative to immunoassay detection methods or prespecified peptide ions measured by multiple reaction monitoring (MRM) approaches. Here we describe an evaluation of different methods to post-process peptide level quantification information to support protein level inference. We characterize the methods by examining their ability to recover a known dilution of a standard protein in background matrices of varying complexity. Additionally, the MS1 quantification results are compared to a standard, targeted, MRM approach on the same samples under equivalent instrument conditions. We show the existence of multiple peptides with MS1 quantification sensitivity similar to the best MRM peptides for each of the background matrices studied. Based on these results we provide recommendations on preferred approaches to leveraging quantitative measurements of multiple peptides to improve protein level inference.</p>","PeriodicalId":73474,"journal":{"name":"International journal of proteomics","volume":"2013 ","pages":"674282"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2013/674282","citationCount":"23","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International journal of proteomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/674282","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/4/23 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 23
Abstract
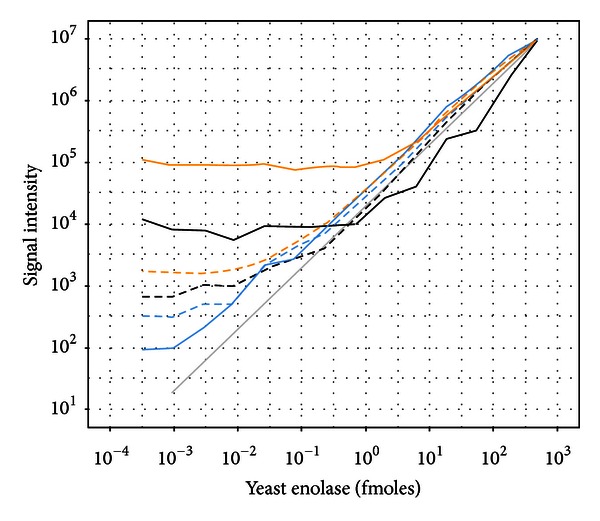
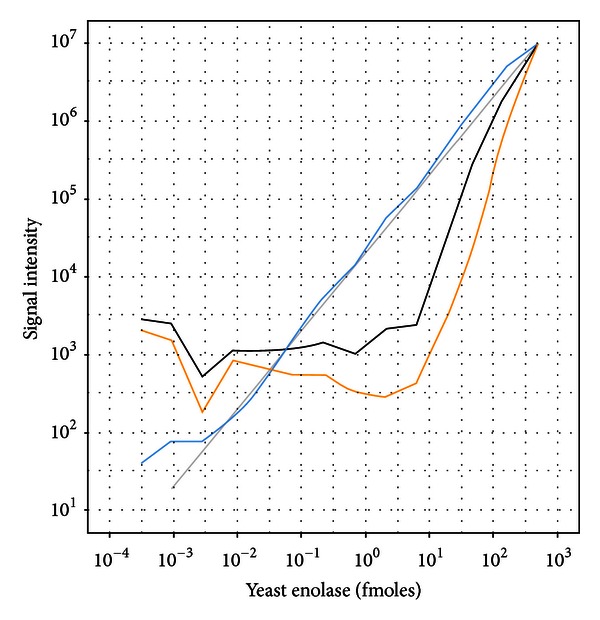
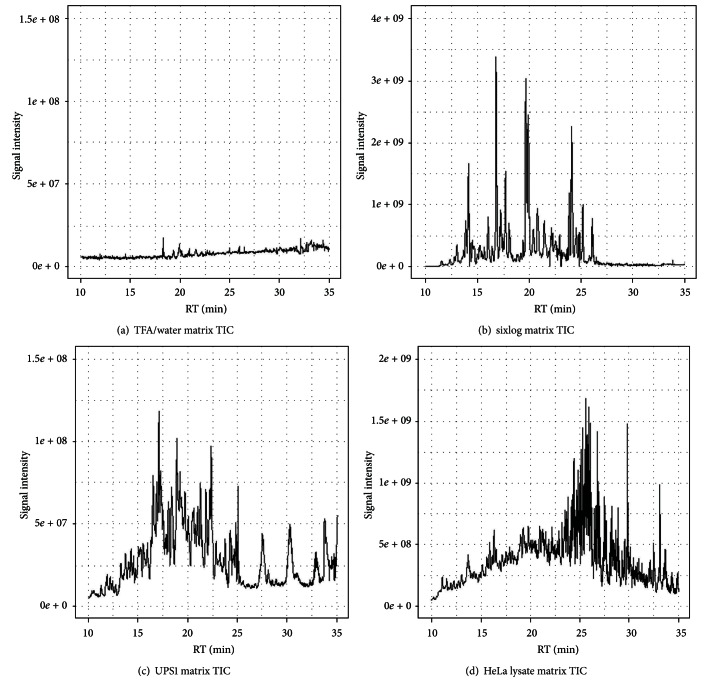
Recent improvements in the mass accuracy and resolution of mass spectrometers have led to renewed interest in label-free quantification using data from the primary mass spectrum (MS1) acquired from data-dependent proteomics experiments. The capacity for higher specificity quantification of peptides from samples enriched for proteins of biological interest offers distinct advantages for hypothesis generating experiments relative to immunoassay detection methods or prespecified peptide ions measured by multiple reaction monitoring (MRM) approaches. Here we describe an evaluation of different methods to post-process peptide level quantification information to support protein level inference. We characterize the methods by examining their ability to recover a known dilution of a standard protein in background matrices of varying complexity. Additionally, the MS1 quantification results are compared to a standard, targeted, MRM approach on the same samples under equivalent instrument conditions. We show the existence of multiple peptides with MS1 quantification sensitivity similar to the best MRM peptides for each of the background matrices studied. Based on these results we provide recommendations on preferred approaches to leveraging quantitative measurements of multiple peptides to improve protein level inference.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
