{"title":"Analysis of gene network robustness based on saturated fixed point attractors.","authors":"Genyuan Li, Herschel Rabitz","doi":"10.1186/1687-4153-2014-4","DOIUrl":null,"url":null,"abstract":"<p><p>The analysis of gene network robustness to noise and mutation is important for fundamental and practical reasons. Robustness refers to the stability of the equilibrium expression state of a gene network to variations of the initial expression state and network topology. Numerical simulation of these variations is commonly used for the assessment of robustness. Since there exists a great number of possible gene network topologies and initial states, even millions of simulations may be still too small to give reliable results. When the initial and equilibrium expression states are restricted to being saturated (i.e., their elements can only take values 1 or -1 corresponding to maximum activation and maximum repression of genes), an analytical gene network robustness assessment is possible. We present this analytical treatment based on determination of the saturated fixed point attractors for sigmoidal function models. The analysis can determine (a) for a given network, which and how many saturated equilibrium states exist and which and how many saturated initial states converge to each of these saturated equilibrium states and (b) for a given saturated equilibrium state or a given pair of saturated equilibrium and initial states, which and how many gene networks, referred to as viable, share this saturated equilibrium state or the pair of saturated equilibrium and initial states. We also show that the viable networks sharing a given saturated equilibrium state must follow certain patterns. These capabilities of the analytical treatment make it possible to properly define and accurately determine robustness to noise and mutation for gene networks. Previous network research conclusions drawn from performing millions of simulations follow directly from the results of our analytical treatment. Furthermore, the analytical results provide criteria for the identification of model validity and suggest modified models of gene network dynamics. The yeast cell-cycle network is used as an illustration of the practical application of this analytical treatment. </p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2014 1","pages":"4"},"PeriodicalIF":0.0000,"publicationDate":"2014-03-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1687-4153-2014-4","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2014-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 5
Abstract
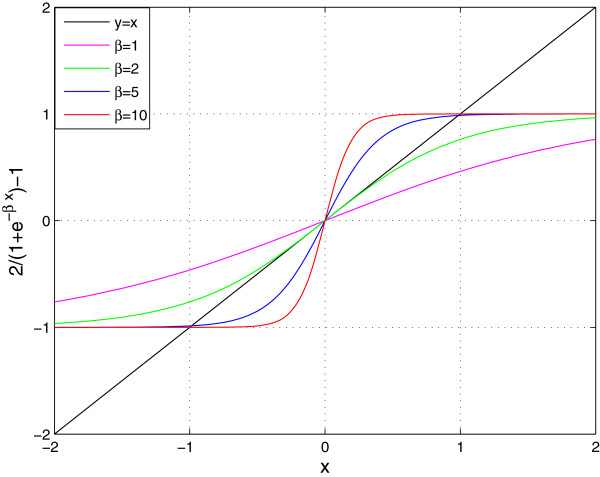
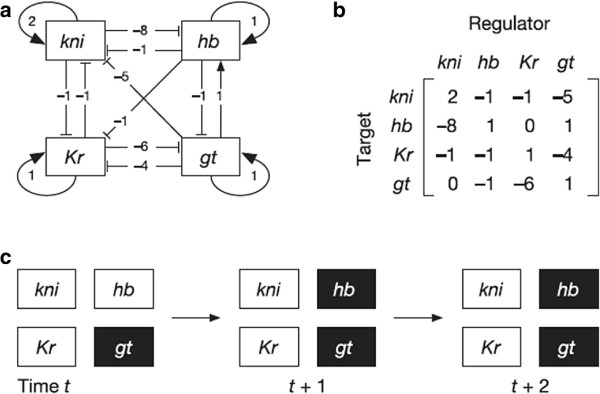
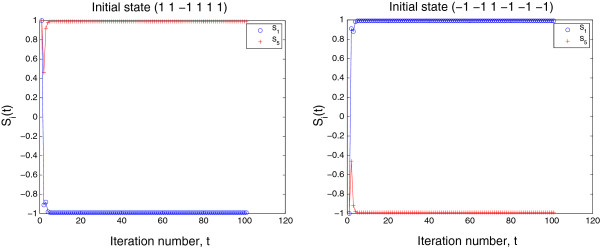
The analysis of gene network robustness to noise and mutation is important for fundamental and practical reasons. Robustness refers to the stability of the equilibrium expression state of a gene network to variations of the initial expression state and network topology. Numerical simulation of these variations is commonly used for the assessment of robustness. Since there exists a great number of possible gene network topologies and initial states, even millions of simulations may be still too small to give reliable results. When the initial and equilibrium expression states are restricted to being saturated (i.e., their elements can only take values 1 or -1 corresponding to maximum activation and maximum repression of genes), an analytical gene network robustness assessment is possible. We present this analytical treatment based on determination of the saturated fixed point attractors for sigmoidal function models. The analysis can determine (a) for a given network, which and how many saturated equilibrium states exist and which and how many saturated initial states converge to each of these saturated equilibrium states and (b) for a given saturated equilibrium state or a given pair of saturated equilibrium and initial states, which and how many gene networks, referred to as viable, share this saturated equilibrium state or the pair of saturated equilibrium and initial states. We also show that the viable networks sharing a given saturated equilibrium state must follow certain patterns. These capabilities of the analytical treatment make it possible to properly define and accurately determine robustness to noise and mutation for gene networks. Previous network research conclusions drawn from performing millions of simulations follow directly from the results of our analytical treatment. Furthermore, the analytical results provide criteria for the identification of model validity and suggest modified models of gene network dynamics. The yeast cell-cycle network is used as an illustration of the practical application of this analytical treatment.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
