Alexandros Iliadis, Dimitris Anastassiou, Xiaodong Wang
{"title":"A sequential Monte Carlo framework for haplotype inference in CNV/SNP genotype data.","authors":"Alexandros Iliadis, Dimitris Anastassiou, Xiaodong Wang","doi":"10.1186/1687-4153-2014-7","DOIUrl":null,"url":null,"abstract":"<p><p>Copy number variations (CNVs) are abundant in the human genome. They have been associated with complex traits in genome-wide association studies (GWAS) and expected to continue playing an important role in identifying the etiology of disease phenotypes. As a result of current high throughput whole-genome single-nucleotide polymorphism (SNP) arrays, we currently have datasets that simultaneously have integer copy numbers in CNV regions as well as SNP genotypes. At the same time, haplotypes that have been shown to offer advantages over genotypes in identifying disease traits even though available for SNP genotypes are largely not available for CNV/SNP data due to insufficient computational tools. We introduce a new framework for inferring haplotypes in CNV/SNP data using a sequential Monte Carlo sampling scheme 'Tree-Based Deterministic Sampling CNV' (TDSCNV). We compare our method with polyHap(v2.0), the only currently available software able to perform inference in CNV/SNP genotypes, on datasets of varying number of markers. We have found that both algorithms show similar accuracy but TDSCNV is an order of magnitude faster while scaling linearly with the number of markers and number of individuals and thus could be the method of choice for haplotype inference in such datasets. Our method is implemented in the TDSCNV package which is available for download at http://www.ee.columbia.edu/~anastas/tdscnv. </p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2014 1","pages":"7"},"PeriodicalIF":0.0000,"publicationDate":"2014-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1687-4153-2014-7","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2014-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/4/24 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 2
Abstract
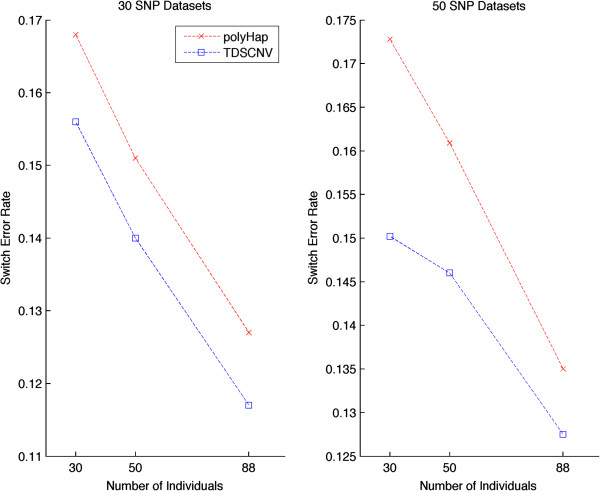
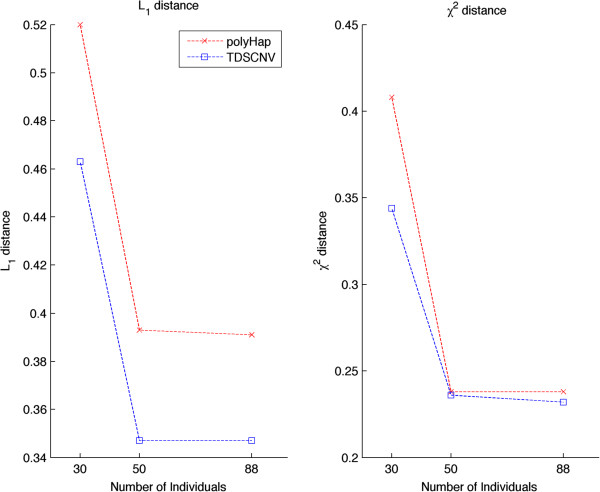
Copy number variations (CNVs) are abundant in the human genome. They have been associated with complex traits in genome-wide association studies (GWAS) and expected to continue playing an important role in identifying the etiology of disease phenotypes. As a result of current high throughput whole-genome single-nucleotide polymorphism (SNP) arrays, we currently have datasets that simultaneously have integer copy numbers in CNV regions as well as SNP genotypes. At the same time, haplotypes that have been shown to offer advantages over genotypes in identifying disease traits even though available for SNP genotypes are largely not available for CNV/SNP data due to insufficient computational tools. We introduce a new framework for inferring haplotypes in CNV/SNP data using a sequential Monte Carlo sampling scheme 'Tree-Based Deterministic Sampling CNV' (TDSCNV). We compare our method with polyHap(v2.0), the only currently available software able to perform inference in CNV/SNP genotypes, on datasets of varying number of markers. We have found that both algorithms show similar accuracy but TDSCNV is an order of magnitude faster while scaling linearly with the number of markers and number of individuals and thus could be the method of choice for haplotype inference in such datasets. Our method is implemented in the TDSCNV package which is available for download at http://www.ee.columbia.edu/~anastas/tdscnv.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
