Shea N Gardner, Crystal J Jaing, Maher M Elsheikh, José Peña, David A Hysom, Monica K Borucki
{"title":"Multiplex degenerate primer design for targeted whole genome amplification of many viral genomes.","authors":"Shea N Gardner, Crystal J Jaing, Maher M Elsheikh, José Peña, David A Hysom, Monica K Borucki","doi":"10.1155/2014/101894","DOIUrl":null,"url":null,"abstract":"<p><p>Background. Targeted enrichment improves coverage of highly mutable viruses at low concentration in complex samples. Degenerate primers that anneal to conserved regions can facilitate amplification of divergent, low concentration variants, even when the strain present is unknown. Results. A tool for designing multiplex sets of degenerate sequencing primers to tile overlapping amplicons across multiple whole genomes is described. The new script, run_tiled_primers, is part of the PriMux software. Primers were designed for each segment of South American hemorrhagic fever viruses, tick-borne encephalitis, Henipaviruses, Arenaviruses, Filoviruses, Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus, and Japanese encephalitis virus. Each group is highly diverse with as little as 5% genome consensus. Primer sets were computationally checked for nontarget cross reactions against the NCBI nucleotide sequence database. Primers for murine hepatitis virus were demonstrated in the lab to specifically amplify selected genes from a laboratory cultured strain that had undergone extensive passage in vitro and in vivo. Conclusions. This software should help researchers design multiplex sets of primers for targeted whole genome enrichment prior to sequencing to obtain better coverage of low titer, divergent viruses. Applications include viral discovery from a complex background and improved sensitivity and coverage of rapidly evolving strains or variants in a gene family. </p>","PeriodicalId":39059,"journal":{"name":"Advances in Bioinformatics","volume":"2014 ","pages":"101894"},"PeriodicalIF":0.0000,"publicationDate":"2014-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2014/101894","citationCount":"13","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances in Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2014/101894","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/8/3 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 13
Abstract
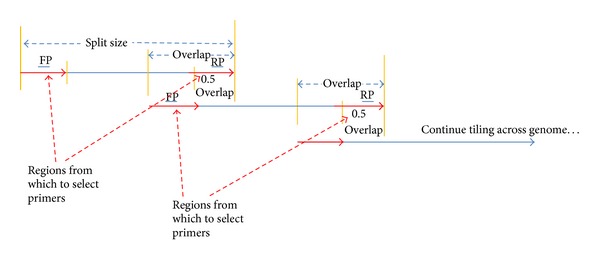
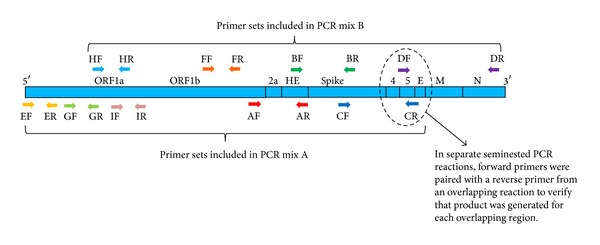
Background. Targeted enrichment improves coverage of highly mutable viruses at low concentration in complex samples. Degenerate primers that anneal to conserved regions can facilitate amplification of divergent, low concentration variants, even when the strain present is unknown. Results. A tool for designing multiplex sets of degenerate sequencing primers to tile overlapping amplicons across multiple whole genomes is described. The new script, run_tiled_primers, is part of the PriMux software. Primers were designed for each segment of South American hemorrhagic fever viruses, tick-borne encephalitis, Henipaviruses, Arenaviruses, Filoviruses, Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus, and Japanese encephalitis virus. Each group is highly diverse with as little as 5% genome consensus. Primer sets were computationally checked for nontarget cross reactions against the NCBI nucleotide sequence database. Primers for murine hepatitis virus were demonstrated in the lab to specifically amplify selected genes from a laboratory cultured strain that had undergone extensive passage in vitro and in vivo. Conclusions. This software should help researchers design multiplex sets of primers for targeted whole genome enrichment prior to sequencing to obtain better coverage of low titer, divergent viruses. Applications include viral discovery from a complex background and improved sensitivity and coverage of rapidly evolving strains or variants in a gene family.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
