{"title":"AdmixKJump: identifying population structure in recently diverged groups.","authors":"Timothy D O'Connor","doi":"10.1186/s13029-014-0031-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Correctly modeling population structure is important for understanding recent evolution and for association studies in humans. While pre-existing knowledge of population history can be used to specify expected levels of subdivision, objective metrics to detect population structure are important and may even be preferable for identifying groups in some situations. One such metric for genomic scale data is implemented in the cross-validation procedure of the program ADMIXTURE, but it has not been evaluated on recently diverged and potentially cryptic levels of population structure. Here, I develop a new method, AdmixKJump, and test both metrics under this scenario.</p><p><strong>Findings: </strong>I show that AdmixKJump is more sensitive to recent population divisions compared to the cross-validation metric using both realistic simulations, as well as 1000 Genomes Project European genomic data. With two populations of 50 individuals each, AdmixKJump is able to detect two populations with 100% accuracy that split at least 10KYA, whereas cross-validation obtains this 100% level at 14KYA. I also show that AdmixKJump is more accurate with fewer samples per population. Furthermore, in contrast to the cross-validation approach, AdmixKJump is able to detect the population split between the Finnish and Tuscan populations of the 1000 Genomes Project.</p><p><strong>Conclusion: </strong>AdmixKJump has more power to detect the number of populations in a cohort of samples with smaller sample sizes and shorter divergence times.</p><p><strong>Availability: </strong>A java implementation can be found at https://sites.google.com/site/igsevolgenomicslab/home/downloads.</p>","PeriodicalId":35052,"journal":{"name":"Source Code for Biology and Medicine","volume":"10 1","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2015-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13029-014-0031-1","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Source Code for Biology and Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13029-014-0031-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Decision Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: Correctly modeling population structure is important for understanding recent evolution and for association studies in humans. While pre-existing knowledge of population history can be used to specify expected levels of subdivision, objective metrics to detect population structure are important and may even be preferable for identifying groups in some situations. One such metric for genomic scale data is implemented in the cross-validation procedure of the program ADMIXTURE, but it has not been evaluated on recently diverged and potentially cryptic levels of population structure. Here, I develop a new method, AdmixKJump, and test both metrics under this scenario.
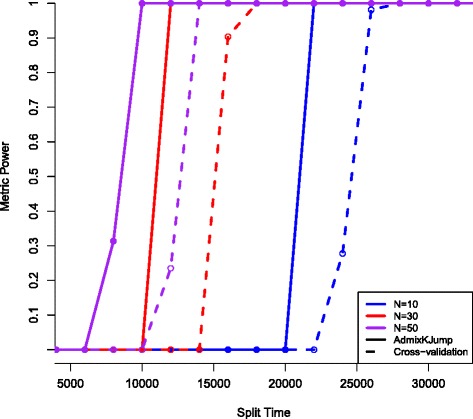
Findings: I show that AdmixKJump is more sensitive to recent population divisions compared to the cross-validation metric using both realistic simulations, as well as 1000 Genomes Project European genomic data. With two populations of 50 individuals each, AdmixKJump is able to detect two populations with 100% accuracy that split at least 10KYA, whereas cross-validation obtains this 100% level at 14KYA. I also show that AdmixKJump is more accurate with fewer samples per population. Furthermore, in contrast to the cross-validation approach, AdmixKJump is able to detect the population split between the Finnish and Tuscan populations of the 1000 Genomes Project.
Conclusion: AdmixKJump has more power to detect the number of populations in a cohort of samples with smaller sample sizes and shorter divergence times.
Availability: A java implementation can be found at https://sites.google.com/site/igsevolgenomicslab/home/downloads.
期刊介绍:
Source Code for Biology and Medicine is a peer-reviewed open access, online journal that publishes articles on source code employed over a wide range of applications in biology and medicine. The journal"s aim is to publish source code for distribution and use in the public domain in order to advance biological and medical research. Through this dissemination, it may be possible to shorten the time required for solving certain computational problems for which there is limited source code availability or resources.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
