{"title":"HMEC: A Heuristic Algorithm for Individual Haplotyping with Minimum Error Correction.","authors":"Md Shamsuzzoha Bayzid, Md Maksudul Alam, Abdullah Mueen, Md Saidur Rahman","doi":"10.1155/2013/291741","DOIUrl":null,"url":null,"abstract":"<p><p>Haplotype is a pattern of single nucleotide polymorphisms (SNPs) on a single chromosome. Constructing a pair of haplotypes from aligned and overlapping but intermixed and erroneous fragments of the chromosomal sequences is a nontrivial problem. Minimum error correction approach aims to minimize the number of errors to be corrected so that the pair of haplotypes can be constructed through consensus of the fragments. We give a heuristic algorithm (HMEC) that searches through alternative solutions using a gain measure and stops whenever no better solution can be achieved. Time complexity of each iteration is O(m (3) k) for an m × k SNP matrix where m and k are the number of fragments (number of rows) and number of SNP sites (number of columns), respectively, in an SNP matrix. Alternative gain measure is also given to reduce running time. We have compared our algorithm with other methods in terms of accuracy and running time on both simulated and real data, and our extensive experimental results indicate the superiority of our algorithm over others. </p>","PeriodicalId":90877,"journal":{"name":"ISRN bioinformatics","volume":"2013 ","pages":"291741"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2013/291741","citationCount":"8","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/291741","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 8
Abstract
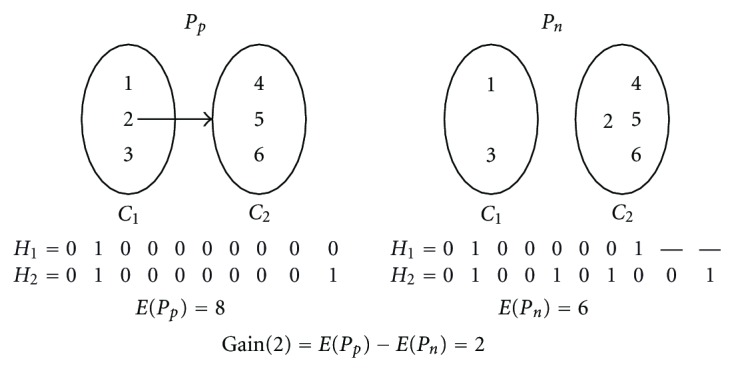
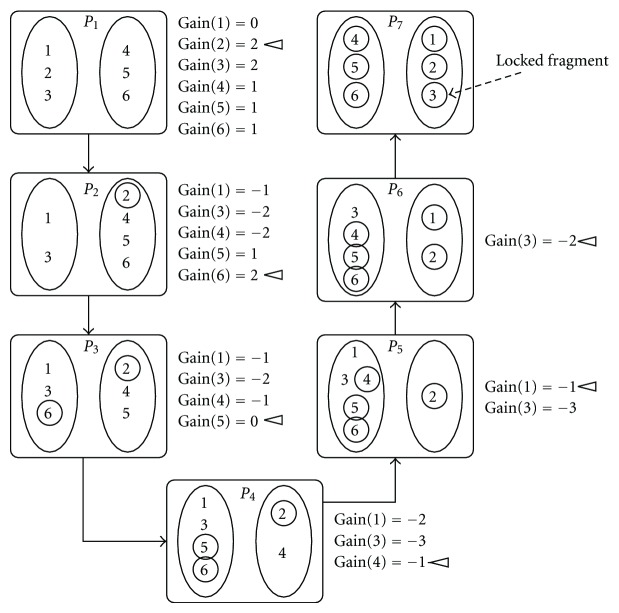
Haplotype is a pattern of single nucleotide polymorphisms (SNPs) on a single chromosome. Constructing a pair of haplotypes from aligned and overlapping but intermixed and erroneous fragments of the chromosomal sequences is a nontrivial problem. Minimum error correction approach aims to minimize the number of errors to be corrected so that the pair of haplotypes can be constructed through consensus of the fragments. We give a heuristic algorithm (HMEC) that searches through alternative solutions using a gain measure and stops whenever no better solution can be achieved. Time complexity of each iteration is O(m (3) k) for an m × k SNP matrix where m and k are the number of fragments (number of rows) and number of SNP sites (number of columns), respectively, in an SNP matrix. Alternative gain measure is also given to reduce running time. We have compared our algorithm with other methods in terms of accuracy and running time on both simulated and real data, and our extensive experimental results indicate the superiority of our algorithm over others.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
