Catarina A Marques, Nicholas J Dickens, Daniel Paape, Samantha J Campbell, Richard McCulloch
{"title":"Genome-wide mapping reveals single-origin chromosome replication in Leishmania, a eukaryotic microbe.","authors":"Catarina A Marques, Nicholas J Dickens, Daniel Paape, Samantha J Campbell, Richard McCulloch","doi":"10.1186/s13059-015-0788-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>DNA replication initiates on defined genome sites, termed origins. Origin usage appears to follow common rules in the eukaryotic organisms examined to date: all chromosomes are replicated from multiple origins, which display variations in firing efficiency and are selected from a larger pool of potential origins. To ask if these features of DNA replication are true of all eukaryotes, we describe genome-wide origin mapping in the parasite Leishmania.</p><p><strong>Results: </strong>Origin mapping in Leishmania suggests a striking divergence in origin usage relative to characterized eukaryotes, since each chromosome appears to be replicated from a single origin. By comparing two species of Leishmania, we find evidence that such origin singularity is maintained in the face of chromosome fusion or fission events during evolution. Mapping Leishmania origins suggests that all origins fire with equal efficiency, and that the genomic sites occupied by origins differ from related non-origins sites. Finally, we provide evidence that origin location in Leishmania displays striking conservation with Trypanosoma brucei, despite the latter parasite replicating its chromosomes from multiple, variable strength origins.</p><p><strong>Conclusions: </strong>The demonstration of chromosome replication for a single origin in Leishmania, a microbial eukaryote, has implications for the evolution of origin multiplicity and associated controls, and may explain the pervasive aneuploidy that characterizes Leishmania chromosome architecture.</p>","PeriodicalId":48922,"journal":{"name":"Genome Biology","volume":"16 ","pages":"230"},"PeriodicalIF":12.3000,"publicationDate":"2015-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4612428/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13059-015-0788-9","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
Background: DNA replication initiates on defined genome sites, termed origins. Origin usage appears to follow common rules in the eukaryotic organisms examined to date: all chromosomes are replicated from multiple origins, which display variations in firing efficiency and are selected from a larger pool of potential origins. To ask if these features of DNA replication are true of all eukaryotes, we describe genome-wide origin mapping in the parasite Leishmania.
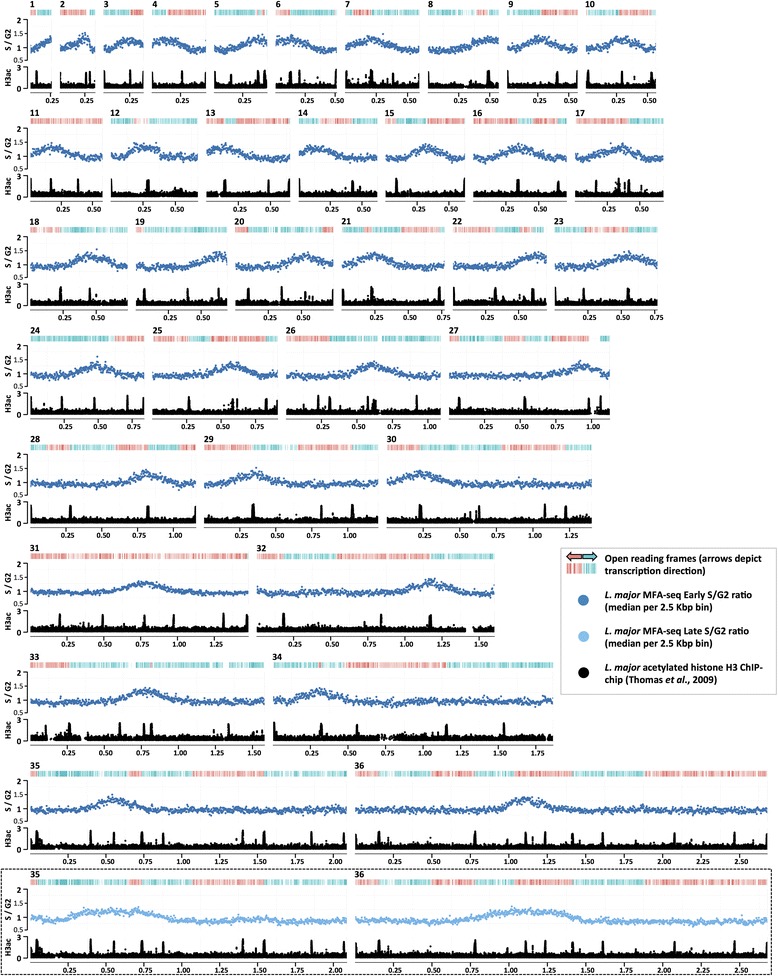
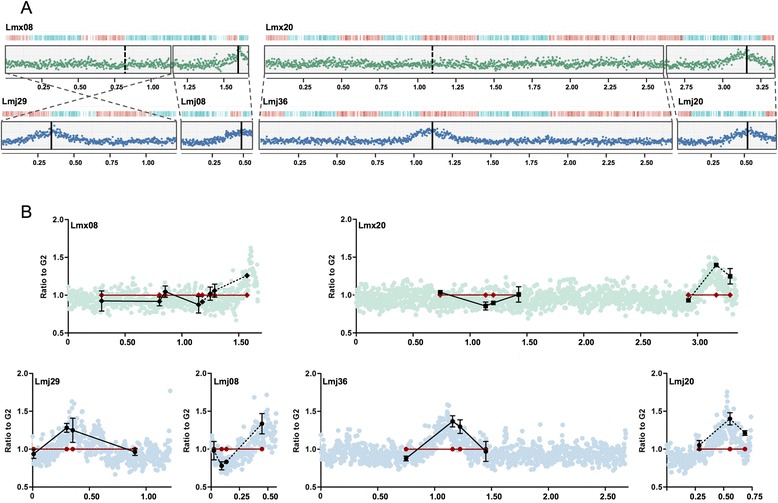
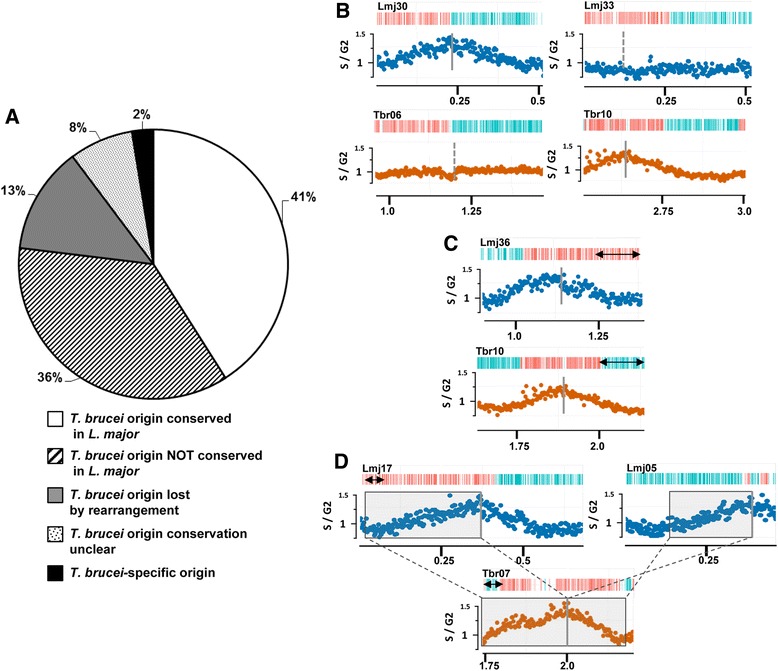
Results: Origin mapping in Leishmania suggests a striking divergence in origin usage relative to characterized eukaryotes, since each chromosome appears to be replicated from a single origin. By comparing two species of Leishmania, we find evidence that such origin singularity is maintained in the face of chromosome fusion or fission events during evolution. Mapping Leishmania origins suggests that all origins fire with equal efficiency, and that the genomic sites occupied by origins differ from related non-origins sites. Finally, we provide evidence that origin location in Leishmania displays striking conservation with Trypanosoma brucei, despite the latter parasite replicating its chromosomes from multiple, variable strength origins.
Conclusions: The demonstration of chromosome replication for a single origin in Leishmania, a microbial eukaryote, has implications for the evolution of origin multiplicity and associated controls, and may explain the pervasive aneuploidy that characterizes Leishmania chromosome architecture.
背景:DNA 复制始于确定的基因组位点,即起源。起源的使用似乎遵循迄今为止所研究的真核生物的共同规则:所有染色体都由多个起源复制,这些起源的点火效率各不相同,而且是从更大的潜在起源库中挑选出来的。为了弄清 DNA 复制的这些特征是否适用于所有真核生物,我们描述了利什曼原虫的全基因组起源图谱:利什曼原虫的原点图谱表明,与特征真核生物相比,原点的使用存在显著差异,因为每条染色体似乎都是从一个原点复制的。通过比较利什曼原虫的两个物种,我们发现有证据表明,面对进化过程中的染色体融合或裂变事件,这种起源的单一性得以保持。绘制利什曼原种的图谱表明,所有原种都能以相同的效率起火,而且原种占据的基因组位点与相关的非原种位点不同。最后,我们提供的证据表明,利什曼原虫的起源位置与布氏锥虫显示出惊人的一致性,尽管后者的寄生虫通过多个不同强度的起源复制染色体:结论:利什曼原虫是一种微生物真核生物,它的染色体复制只有一个起源,这对起源多重性和相关控制的进化具有重要意义,也可以解释利什曼原虫染色体结构中普遍存在的非整倍体现象。
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
