{"title":"Hyaluronan Synthase 3 Null Mice Exhibit Decreased Intestinal Inflammation and Tissue Damage in the DSS-Induced Colitis Model.","authors":"Sean P Kessler, Dana R Obery, Carol de la Motte","doi":"10.1155/2015/745237","DOIUrl":null,"url":null,"abstract":"<p><p>Hyaluronan (HA) overproduction is a hallmark of multiple inflammatory diseases, including inflammatory bowel disease (IBD). Hyaluronan can act as a leukocyte recruitment molecule and in the most common mouse model of intestinal inflammation, the chemically induced dextran sodium sulfate (DSS) experimental colitis model, we previously determined that changes in colon distribution of HA occur before inflammation. Therefore, we hypothesized that, during a pathologic challenge, HA promotes inflammation. In this study, we tested the progression of inflammation in mice null for the hyaluronan synthase genes (HAS1, HAS3, or both HAS1 and HAS3) in the DSS-colitis model. Our data demonstrate that both the HAS1/HAS3 double and the HAS3 null mice are protected from colitis, compared to wild-type and HAS1 null mice, as determined by measurement of weight loss, disease activity, serum IL-6 levels, histologic scoring, and immunohistochemistry. Most notable is the dramatic increase in submucosal microvasculature, hyaluronan deposition, and leukocyte infiltration in the inflamed colon tissue of wild-type and HAS1 null mice. Our data suggest, HAS3 plays a crucial role in driving gut inflammation. Developing a temporary targeted therapeutic intervention of HAS3 expression or function in the microcirculation may emerge as a desirable strategy toward tempering colitis in patients undergoing flares of IBD. </p>","PeriodicalId":39084,"journal":{"name":"International Journal of Cell Biology","volume":"2015 ","pages":"745237"},"PeriodicalIF":0.0000,"publicationDate":"2015-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2015/745237","citationCount":"44","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Cell Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2015/745237","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/9/10 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 44
Abstract
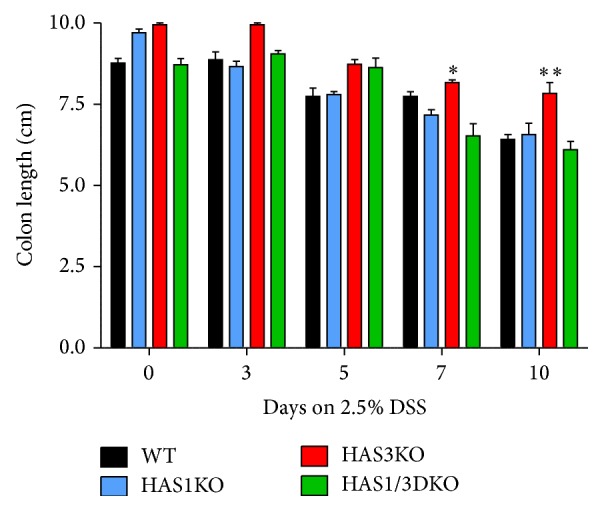
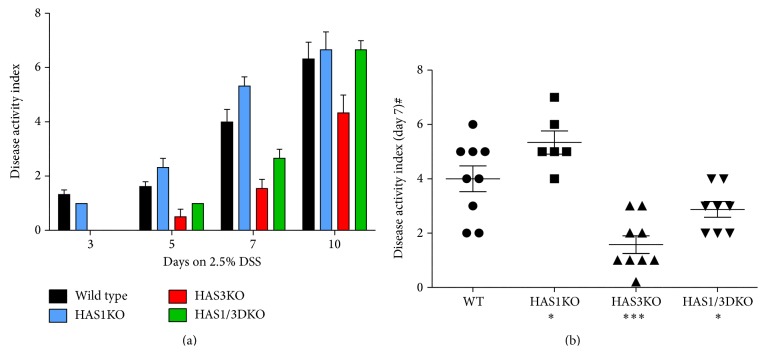
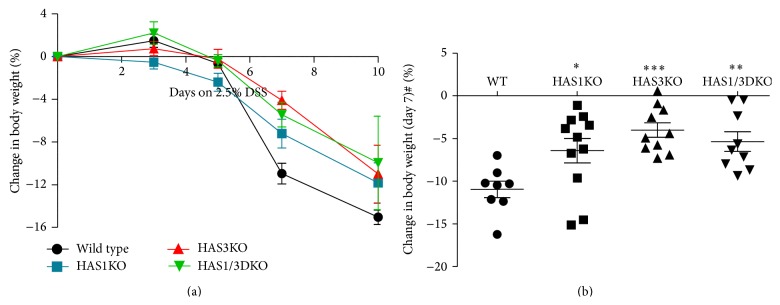
Hyaluronan (HA) overproduction is a hallmark of multiple inflammatory diseases, including inflammatory bowel disease (IBD). Hyaluronan can act as a leukocyte recruitment molecule and in the most common mouse model of intestinal inflammation, the chemically induced dextran sodium sulfate (DSS) experimental colitis model, we previously determined that changes in colon distribution of HA occur before inflammation. Therefore, we hypothesized that, during a pathologic challenge, HA promotes inflammation. In this study, we tested the progression of inflammation in mice null for the hyaluronan synthase genes (HAS1, HAS3, or both HAS1 and HAS3) in the DSS-colitis model. Our data demonstrate that both the HAS1/HAS3 double and the HAS3 null mice are protected from colitis, compared to wild-type and HAS1 null mice, as determined by measurement of weight loss, disease activity, serum IL-6 levels, histologic scoring, and immunohistochemistry. Most notable is the dramatic increase in submucosal microvasculature, hyaluronan deposition, and leukocyte infiltration in the inflamed colon tissue of wild-type and HAS1 null mice. Our data suggest, HAS3 plays a crucial role in driving gut inflammation. Developing a temporary targeted therapeutic intervention of HAS3 expression or function in the microcirculation may emerge as a desirable strategy toward tempering colitis in patients undergoing flares of IBD.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
