Cameron D Fyfe, Rhys Grinter, Inokentijs Josts, Khedidja Mosbahi, Aleksander W Roszak, Richard J Cogdell, Daniel M Wall, Richard J S Burchmore, Olwyn Byron, Daniel Walker
{"title":"Structure of protease-cleaved Escherichia coli α-2-macroglobulin reveals a putative mechanism of conformational activation for protease entrapment.","authors":"Cameron D Fyfe, Rhys Grinter, Inokentijs Josts, Khedidja Mosbahi, Aleksander W Roszak, Richard J Cogdell, Daniel M Wall, Richard J S Burchmore, Olwyn Byron, Daniel Walker","doi":"10.1107/S1399004715008548","DOIUrl":null,"url":null,"abstract":"<p><p>Bacterial α-2-macroglobulins have been suggested to function in defence as broad-spectrum inhibitors of host proteases that breach the outer membrane. Here, the X-ray structure of protease-cleaved Escherichia coli α-2-macroglobulin is described, which reveals a putative mechanism of activation and conformational change essential for protease inhibition. In this competitive mechanism, protease cleavage of the bait-region domain results in the untethering of an intrinsically disordered region of this domain which disrupts native interdomain interactions that maintain E. coli α-2-macroglobulin in the inactivated form. The resulting global conformational change results in entrapment of the protease and activation of the thioester bond that covalently links to the attacking protease. Owing to the similarity in structure and domain architecture of Escherichia coli α-2-macroglobulin and human α-2-macroglobulin, this protease-activation mechanism is likely to operate across the diverse members of this group. </p>","PeriodicalId":7047,"journal":{"name":"Acta crystallographica. Section D, Biological crystallography","volume":" ","pages":"1478-86"},"PeriodicalIF":0.0000,"publicationDate":"2015-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4498604/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta crystallographica. Section D, Biological crystallography","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1107/S1399004715008548","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/6/30 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Bacterial α-2-macroglobulins have been suggested to function in defence as broad-spectrum inhibitors of host proteases that breach the outer membrane. Here, the X-ray structure of protease-cleaved Escherichia coli α-2-macroglobulin is described, which reveals a putative mechanism of activation and conformational change essential for protease inhibition. In this competitive mechanism, protease cleavage of the bait-region domain results in the untethering of an intrinsically disordered region of this domain which disrupts native interdomain interactions that maintain E. coli α-2-macroglobulin in the inactivated form. The resulting global conformational change results in entrapment of the protease and activation of the thioester bond that covalently links to the attacking protease. Owing to the similarity in structure and domain architecture of Escherichia coli α-2-macroglobulin and human α-2-macroglobulin, this protease-activation mechanism is likely to operate across the diverse members of this group.
细菌的α-2-巨球蛋白被认为具有防御功能,是破坏外膜的宿主蛋白酶的广谱抑制剂。本文描述了蛋白酶裂解的大肠杆菌α-2-巨球蛋白的 X 射线结构,揭示了蛋白酶抑制所必需的活化和构象变化的推定机制。在这一竞争机制中,蛋白酶对诱饵区结构域的裂解导致该结构域的一个内在无序区被解开,从而破坏了维持大肠杆菌α-2-巨球蛋白失活形态的原生结构域间相互作用。由此产生的整体构象变化导致蛋白酶被截留,并激活了与攻击蛋白酶共价连接的硫酯键。由于大肠杆菌α-2-巨球蛋白和人类α-2-巨球蛋白在结构和结构域上的相似性,这种蛋白酶活化机制很可能适用于该类蛋白的不同成员。
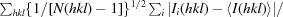


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
