Javier Arsuaga, Tyler Borrman, Raymond Cavalcante, Georgina Gonzalez, Catherine Park
{"title":"Identification of Copy Number Aberrations in Breast Cancer Subtypes Using Persistence Topology.","authors":"Javier Arsuaga, Tyler Borrman, Raymond Cavalcante, Georgina Gonzalez, Catherine Park","doi":"10.3390/microarrays4030339","DOIUrl":null,"url":null,"abstract":"<p><p>DNA copy number aberrations (CNAs) are of biological and medical interest because they help identify regulatory mechanisms underlying tumor initiation and evolution. Identification of tumor-driving CNAs (driver CNAs) however remains a challenging task, because they are frequently hidden by CNAs that are the product of random events that take place during tumor evolution. Experimental detection of CNAs is commonly accomplished through array comparative genomic hybridization (aCGH) assays followed by supervised and/or unsupervised statistical methods that combine the segmented profiles of all patients to identify driver CNAs. Here, we extend a previously-presented supervised algorithm for the identification of CNAs that is based on a topological representation of the data. Our method associates a two-dimensional (2D) point cloud with each aCGH profile and generates a sequence of simplicial complexes, mathematical objects that generalize the concept of a graph. This representation of the data permits segmenting the data at different resolutions and identifying CNAs by interrogating the topological properties of these simplicial complexes. We tested our approach on a published dataset with the goal of identifying specific breast cancer CNAs associated with specific molecular subtypes. Identification of CNAs associated with each subtype was performed by analyzing each subtype separately from the others and by taking the rest of the subtypes as the control. Our results found a new amplification in 11q at the location of the progesterone receptor in the Luminal A subtype. Aberrations in the Luminal B subtype were found only upon removal of the basal-like subtype from the control set. Under those conditions, all regions found in the original publication, except for 17q, were confirmed; all aberrations, except those in chromosome arms 8q and 12q were confirmed in the basal-like subtype. These two chromosome arms, however, were detected only upon removal of three patients with exceedingly large copy number values. More importantly, we detected 10 and 21 additional regions in the Luminal B and basal-like subtypes, respectively. Most of the additional regions were either validated on an independent dataset and/or using GISTIC. Furthermore, we found three new CNAs in the basal-like subtype: a combination of gains and losses in 1p, a gain in 2p and a loss in 14q. Based on these results, we suggest that topological approaches that incorporate multiresolution analyses and that interrogate topological properties of the data can help in the identification of copy number changes in cancer. </p>","PeriodicalId":56355,"journal":{"name":"Microarrays","volume":" ","pages":"339-69"},"PeriodicalIF":0.0000,"publicationDate":"2015-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/microarrays4030339","citationCount":"21","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microarrays","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/microarrays4030339","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 21
Abstract
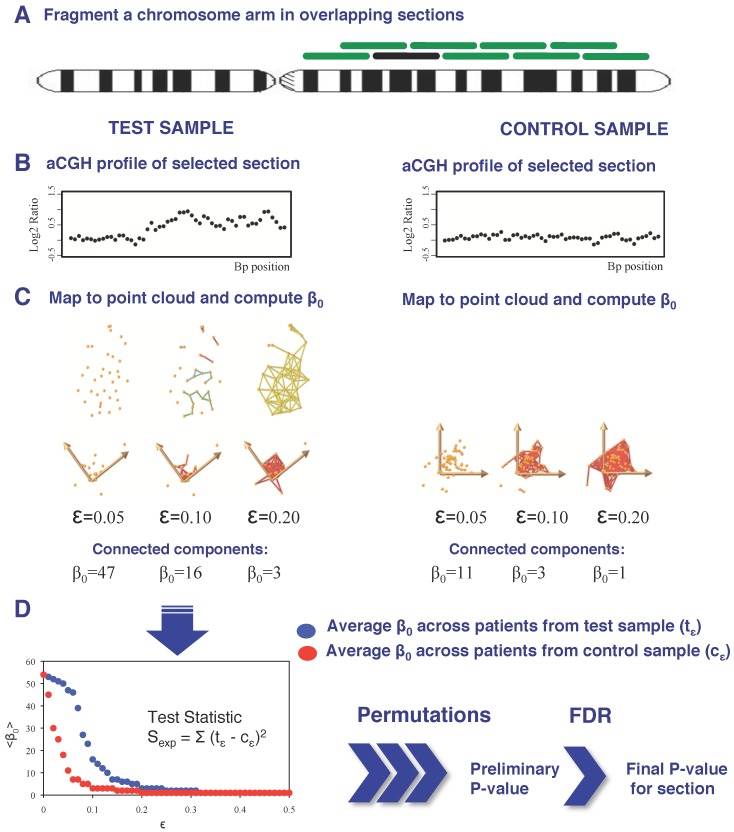
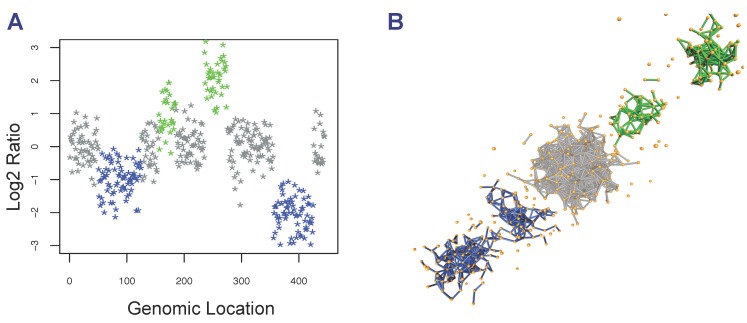
DNA copy number aberrations (CNAs) are of biological and medical interest because they help identify regulatory mechanisms underlying tumor initiation and evolution. Identification of tumor-driving CNAs (driver CNAs) however remains a challenging task, because they are frequently hidden by CNAs that are the product of random events that take place during tumor evolution. Experimental detection of CNAs is commonly accomplished through array comparative genomic hybridization (aCGH) assays followed by supervised and/or unsupervised statistical methods that combine the segmented profiles of all patients to identify driver CNAs. Here, we extend a previously-presented supervised algorithm for the identification of CNAs that is based on a topological representation of the data. Our method associates a two-dimensional (2D) point cloud with each aCGH profile and generates a sequence of simplicial complexes, mathematical objects that generalize the concept of a graph. This representation of the data permits segmenting the data at different resolutions and identifying CNAs by interrogating the topological properties of these simplicial complexes. We tested our approach on a published dataset with the goal of identifying specific breast cancer CNAs associated with specific molecular subtypes. Identification of CNAs associated with each subtype was performed by analyzing each subtype separately from the others and by taking the rest of the subtypes as the control. Our results found a new amplification in 11q at the location of the progesterone receptor in the Luminal A subtype. Aberrations in the Luminal B subtype were found only upon removal of the basal-like subtype from the control set. Under those conditions, all regions found in the original publication, except for 17q, were confirmed; all aberrations, except those in chromosome arms 8q and 12q were confirmed in the basal-like subtype. These two chromosome arms, however, were detected only upon removal of three patients with exceedingly large copy number values. More importantly, we detected 10 and 21 additional regions in the Luminal B and basal-like subtypes, respectively. Most of the additional regions were either validated on an independent dataset and/or using GISTIC. Furthermore, we found three new CNAs in the basal-like subtype: a combination of gains and losses in 1p, a gain in 2p and a loss in 14q. Based on these results, we suggest that topological approaches that incorporate multiresolution analyses and that interrogate topological properties of the data can help in the identification of copy number changes in cancer.
期刊介绍:
High-Throughput (formerly Microarrays, ISSN 2076-3905) is a multidisciplinary peer-reviewed scientific journal that provides an advanced forum for the publication of studies reporting high-dimensional approaches and developments in Life Sciences, Chemistry and related fields. Our aim is to encourage scientists to publish their experimental and theoretical results based on high-throughput techniques as well as computational and statistical tools for data analysis and interpretation. The full experimental or methodological details must be provided so that the results can be reproduced. There is no restriction on the length of the papers. High-Throughput invites submissions covering several topics, including, but not limited to: Microarrays, DNA Sequencing, RNA Sequencing, Protein Identification and Quantification, Cell-based Approaches, Omics Technologies, Imaging, Bioinformatics, Computational Biology/Chemistry, Statistics, Integrative Omics, Drug Discovery and Development, Microfluidics, Lab-on-a-chip, Data Mining, Databases, Multiplex Assays.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
