Mostafa A Salama, Aboul Ella Hassanien, Ahmad Mostafa
{"title":"The prediction of virus mutation using neural networks and rough set techniques.","authors":"Mostafa A Salama, Aboul Ella Hassanien, Ahmad Mostafa","doi":"10.1186/s13637-016-0042-0","DOIUrl":null,"url":null,"abstract":"<p><p>Viral evolution remains to be a main obstacle in the effectiveness of antiviral treatments. The ability to predict this evolution will help in the early detection of drug-resistant strains and will potentially facilitate the design of more efficient antiviral treatments. Various tools has been utilized in genome studies to achieve this goal. One of these tools is machine learning, which facilitates the study of structure-activity relationships, secondary and tertiary structure evolution prediction, and sequence error correction. This work proposes a novel machine learning technique for the prediction of the possible point mutations that appear on alignments of primary RNA sequence structure. It predicts the genotype of each nucleotide in the RNA sequence, and proves that a nucleotide in an RNA sequence changes based on the other nucleotides in the sequence. Neural networks technique is utilized in order to predict new strains, then a rough set theory based algorithm is introduced to extract these point mutation patterns. This algorithm is applied on a number of aligned RNA isolates time-series species of the Newcastle virus. Two different data sets from two sources are used in the validation of these techniques. The results show that the accuracy of this technique in predicting the nucleotides in the new generation is as high as 75 %. The mutation rules are visualized for the analysis of the correlation between different nucleotides in the same RNA sequence.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2016 1","pages":"10"},"PeriodicalIF":0.0000,"publicationDate":"2016-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13637-016-0042-0","citationCount":"28","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13637-016-0042-0","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/12/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 28
Abstract
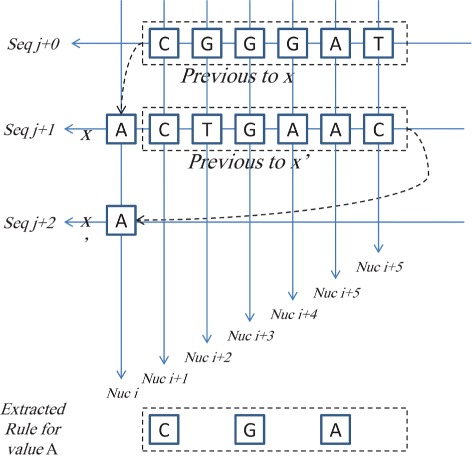
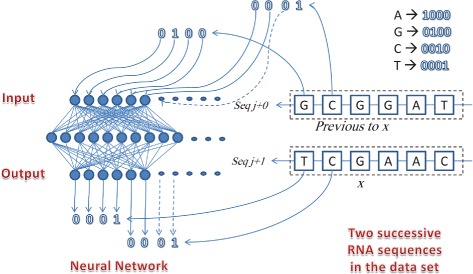
Viral evolution remains to be a main obstacle in the effectiveness of antiviral treatments. The ability to predict this evolution will help in the early detection of drug-resistant strains and will potentially facilitate the design of more efficient antiviral treatments. Various tools has been utilized in genome studies to achieve this goal. One of these tools is machine learning, which facilitates the study of structure-activity relationships, secondary and tertiary structure evolution prediction, and sequence error correction. This work proposes a novel machine learning technique for the prediction of the possible point mutations that appear on alignments of primary RNA sequence structure. It predicts the genotype of each nucleotide in the RNA sequence, and proves that a nucleotide in an RNA sequence changes based on the other nucleotides in the sequence. Neural networks technique is utilized in order to predict new strains, then a rough set theory based algorithm is introduced to extract these point mutation patterns. This algorithm is applied on a number of aligned RNA isolates time-series species of the Newcastle virus. Two different data sets from two sources are used in the validation of these techniques. The results show that the accuracy of this technique in predicting the nucleotides in the new generation is as high as 75 %. The mutation rules are visualized for the analysis of the correlation between different nucleotides in the same RNA sequence.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
