{"title":"Clinical features and genotype-phenotype correlation analysis in patients with <i>ATL1</i> mutations: A literature reanalysis.","authors":"Guo-Hua Zhao, Xiao-Min Liu","doi":"10.1186/s40035-017-0079-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The hereditary spastic paraplegias (HSPs) are a group of clinically and genetically heterogeneous disorders. Approximately 10% of the autosomal dominant (AD) HSPs (ADHSPs) have the spastic paraplegia 3A (SPG3A) genotype which is caused by <i>ATL1</i> gene mutations. Currently there are more than 60 reported <i>ATL1</i> gene mutations and the genotype-phenotype correlation remains unclear. The study aims to investigate the genotype-phenotype correlation in SPG3A patients.</p><p><strong>Methods: </strong>We performed a reanalysis of the clinical features and genotype-phenotype correlations in 51 reported studies exhibiting an <i>ATL1</i> gene mutation.</p><p><strong>Results: </strong>Most HSPs-SPG3A patients exhibited an early age at onset (AAO) of <10 years old, and showed an autosomal dominant pure spastic paraplegia. We found that 14% of the HSPs-SPG3A patients presented complicated phenotypes, with distal atrophy being the most common complicated symptom. The AAO of each mutation group was not statistically significant (<i>P</i> > 0.05). The mutational spectrum associated with <i>ATL1</i> gene mutation is wide, and most mutations are missense mutations, but do not involve the functional motif of <i>ATL1</i> gene encoded atlastin-1 protein.</p><p><strong>Conclusions: </strong>Our findings indicate that there is no clear genotype-phenotype correlation in HSPs-SPG3A patients. We also find that exons 4, 7, 8 and 12 are mutation hotspots in <i>ATL1</i> gene.</p>","PeriodicalId":23269,"journal":{"name":"Translational Neurodegeneration","volume":"6 ","pages":"9"},"PeriodicalIF":15.2000,"publicationDate":"2017-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40035-017-0079-3","citationCount":"26","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational Neurodegeneration","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40035-017-0079-3","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2017/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 26
Abstract
Background: The hereditary spastic paraplegias (HSPs) are a group of clinically and genetically heterogeneous disorders. Approximately 10% of the autosomal dominant (AD) HSPs (ADHSPs) have the spastic paraplegia 3A (SPG3A) genotype which is caused by ATL1 gene mutations. Currently there are more than 60 reported ATL1 gene mutations and the genotype-phenotype correlation remains unclear. The study aims to investigate the genotype-phenotype correlation in SPG3A patients.
Methods: We performed a reanalysis of the clinical features and genotype-phenotype correlations in 51 reported studies exhibiting an ATL1 gene mutation.
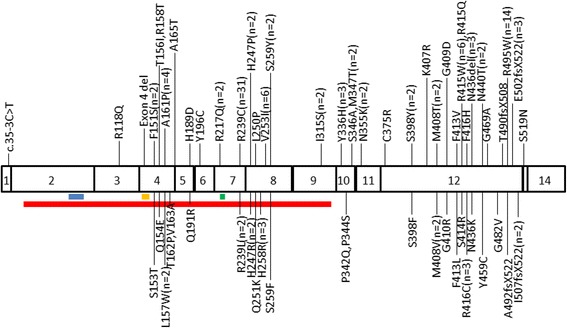
Results: Most HSPs-SPG3A patients exhibited an early age at onset (AAO) of <10 years old, and showed an autosomal dominant pure spastic paraplegia. We found that 14% of the HSPs-SPG3A patients presented complicated phenotypes, with distal atrophy being the most common complicated symptom. The AAO of each mutation group was not statistically significant (P > 0.05). The mutational spectrum associated with ATL1 gene mutation is wide, and most mutations are missense mutations, but do not involve the functional motif of ATL1 gene encoded atlastin-1 protein.
Conclusions: Our findings indicate that there is no clear genotype-phenotype correlation in HSPs-SPG3A patients. We also find that exons 4, 7, 8 and 12 are mutation hotspots in ATL1 gene.
期刊介绍:
Translational Neurodegeneration, an open-access, peer-reviewed journal, addresses all aspects of neurodegenerative diseases. It serves as a prominent platform for research, therapeutics, and education, fostering discussions and insights across basic, translational, and clinical research domains. Covering Parkinson's disease, Alzheimer's disease, and other neurodegenerative conditions, it welcomes contributions on epidemiology, pathogenesis, diagnosis, prevention, drug development, rehabilitation, and drug delivery. Scientists, clinicians, and physician-scientists are encouraged to share their work in this specialized journal tailored to their fields.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
