{"title":"Applying Expression Profile Similarity for Discovery of Patient-Specific Functional Mutations.","authors":"Guofeng Meng","doi":"10.3390/ht7010006","DOIUrl":null,"url":null,"abstract":"<p><p>The progress of cancer genome sequencing projects yields unprecedented information of mutations for numerous patients. However, the complexity of mutation profiles of cancer patients hinders the further understanding to mechanisms of oncogenesis. One basic question is how to find mutations with functional impacts. In this work, we introduce a computational method to predict functional somatic mutations of each patient by integrating mutation recurrence with expression profile similarity. With this method, the functional mutations are determined by checking the mutation enrichment among a group of patients with similar expression profiles. We applied this method to three cancer types and identified the functional mutations. Comparison of the predictions for three cancer types suggested that most of the functional mutations were cancer-type-specific with one exception to <i>p53</i>. By checking predicted results, we found that our method effectively filtered non-functional mutations resulting from large protein sizes. In addition, this method can also perform functional annotation to each patient to describe their association with signalling pathways or biological processes. In breast cancer, we predicted \"cell adhesion\" and other terms to be significantly associated with oncogenesis.</p>","PeriodicalId":53433,"journal":{"name":"High-Throughput","volume":"7 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2018-02-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/ht7010006","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"High-Throughput","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ht7010006","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 2
Abstract
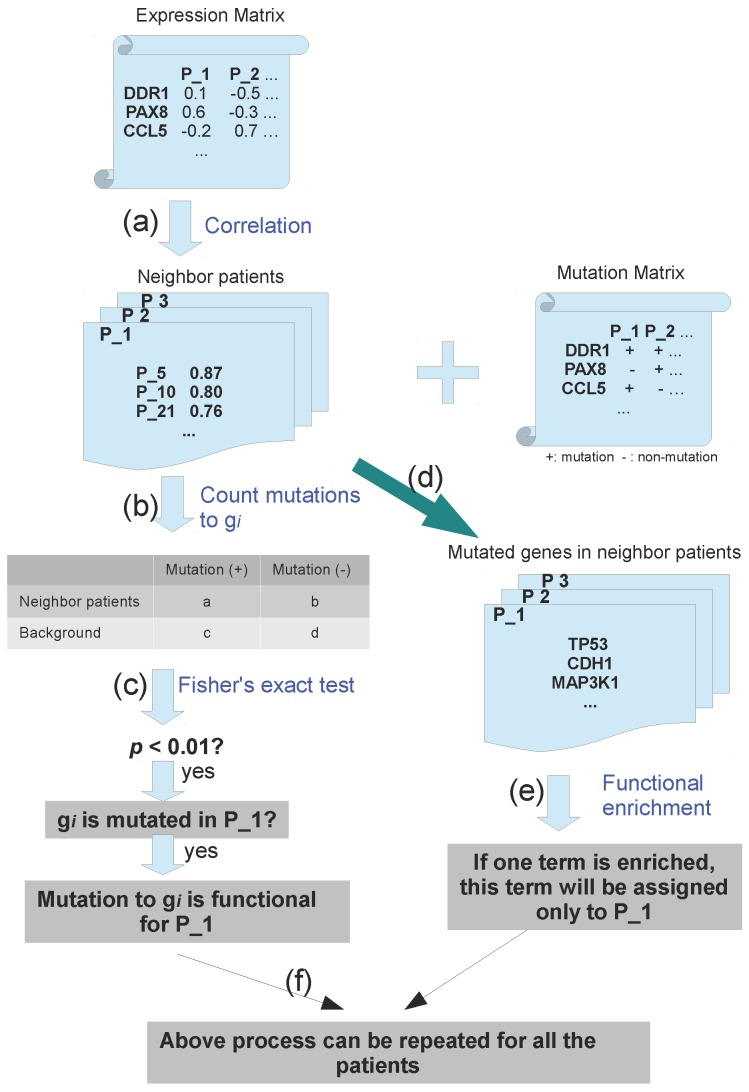
The progress of cancer genome sequencing projects yields unprecedented information of mutations for numerous patients. However, the complexity of mutation profiles of cancer patients hinders the further understanding to mechanisms of oncogenesis. One basic question is how to find mutations with functional impacts. In this work, we introduce a computational method to predict functional somatic mutations of each patient by integrating mutation recurrence with expression profile similarity. With this method, the functional mutations are determined by checking the mutation enrichment among a group of patients with similar expression profiles. We applied this method to three cancer types and identified the functional mutations. Comparison of the predictions for three cancer types suggested that most of the functional mutations were cancer-type-specific with one exception to p53. By checking predicted results, we found that our method effectively filtered non-functional mutations resulting from large protein sizes. In addition, this method can also perform functional annotation to each patient to describe their association with signalling pathways or biological processes. In breast cancer, we predicted "cell adhesion" and other terms to be significantly associated with oncogenesis.
High-ThroughputBiochemistry, Genetics and Molecular Biology-Biotechnology
CiteScore
3.60
自引率
0.00%
发文量
0
审稿时长
9 weeks
期刊介绍:
High-Throughput (formerly Microarrays, ISSN 2076-3905) is a multidisciplinary peer-reviewed scientific journal that provides an advanced forum for the publication of studies reporting high-dimensional approaches and developments in Life Sciences, Chemistry and related fields. Our aim is to encourage scientists to publish their experimental and theoretical results based on high-throughput techniques as well as computational and statistical tools for data analysis and interpretation. The full experimental or methodological details must be provided so that the results can be reproduced. There is no restriction on the length of the papers. High-Throughput invites submissions covering several topics, including, but not limited to: -Microarrays -DNA Sequencing -RNA Sequencing -Protein Identification and Quantification -Cell-based Approaches -Omics Technologies -Imaging -Bioinformatics -Computational Biology/Chemistry -Statistics -Integrative Omics -Drug Discovery and Development -Microfluidics -Lab-on-a-chip -Data Mining -Databases -Multiplex Assays



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
