{"title":"QSAR Modeling and Molecular Docking Analysis of Some Active Compounds against <i>Mycobacterium tuberculosis</i> Receptor (Mtb CYP121).","authors":"Shola Elijah Adeniji, Sani Uba, Adamu Uzairu","doi":"10.1155/2018/1018694","DOIUrl":null,"url":null,"abstract":"<p><p>A quantitative structure-activity relationship (QSAR) study was performed to develop a model that relates the structures of 50 compounds to their activities against <i>M. tuberculosis</i>. The compounds were optimized by employing density functional theory (DFT) with B3LYP/6-31G<sup>⁎</sup>. The Genetic Function Algorithm (GFA) was used to select the descriptors and to generate the correlation model that relates the structural features of the compounds to their biological activities. The optimum model has squared correlation coefficient (<i>R</i><sup>2</sup>) of 0.9202, adjusted squared correlation coefficient (<i>R</i><sub>adj</sub>) of 0.91012, and leave-one-out (LOO) cross-validation coefficient (<i>Q</i><sub>cv</sub><sup>2</sup>) value of 0.8954. The external validation test used for confirming the predictive power of the built model has <i>R</i><sup>2</sup>pred value of 0.8842. These parameters confirm the stability and robustness of the model. Docking analysis showed the best compound with high docking affinity of -14.6 kcal/mol which formed hydrophobic interaction and hydrogen bond with amino acid residues of <i>M. tuberculosis</i> cytochromes (Mtb CYP121). QSAR and molecular docking studies provide valuable approach for pharmaceutical and medicinal chemists to design and synthesize new anti-<i>Mycobacterium tuberculosis</i> compounds.</p>","PeriodicalId":16788,"journal":{"name":"Journal of Pathogens","volume":"2018 ","pages":"1018694"},"PeriodicalIF":1.1000,"publicationDate":"2018-05-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5971244/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Pathogens","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2018/1018694","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
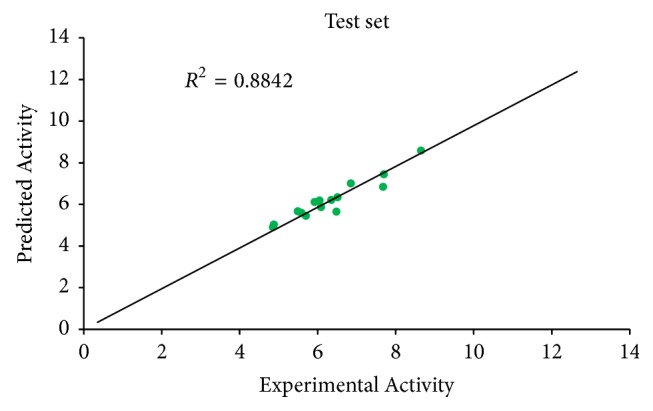
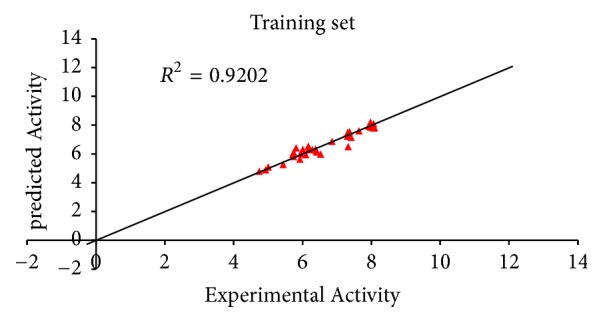
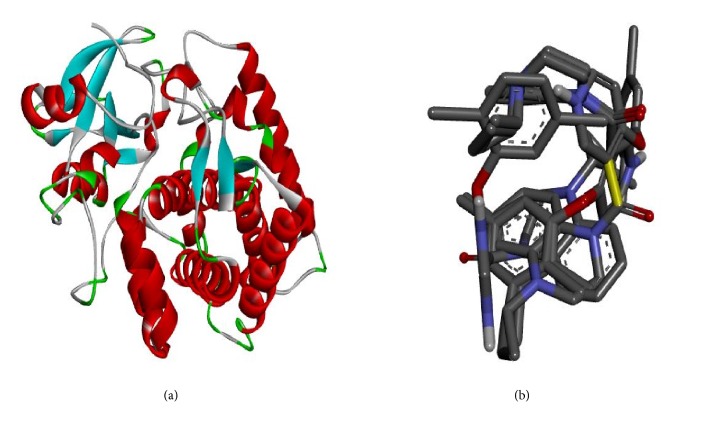
A quantitative structure-activity relationship (QSAR) study was performed to develop a model that relates the structures of 50 compounds to their activities against M. tuberculosis. The compounds were optimized by employing density functional theory (DFT) with B3LYP/6-31G⁎. The Genetic Function Algorithm (GFA) was used to select the descriptors and to generate the correlation model that relates the structural features of the compounds to their biological activities. The optimum model has squared correlation coefficient (R2) of 0.9202, adjusted squared correlation coefficient (Radj) of 0.91012, and leave-one-out (LOO) cross-validation coefficient (Qcv2) value of 0.8954. The external validation test used for confirming the predictive power of the built model has R2pred value of 0.8842. These parameters confirm the stability and robustness of the model. Docking analysis showed the best compound with high docking affinity of -14.6 kcal/mol which formed hydrophobic interaction and hydrogen bond with amino acid residues of M. tuberculosis cytochromes (Mtb CYP121). QSAR and molecular docking studies provide valuable approach for pharmaceutical and medicinal chemists to design and synthesize new anti-Mycobacterium tuberculosis compounds.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
