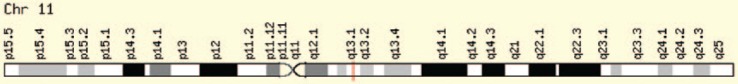
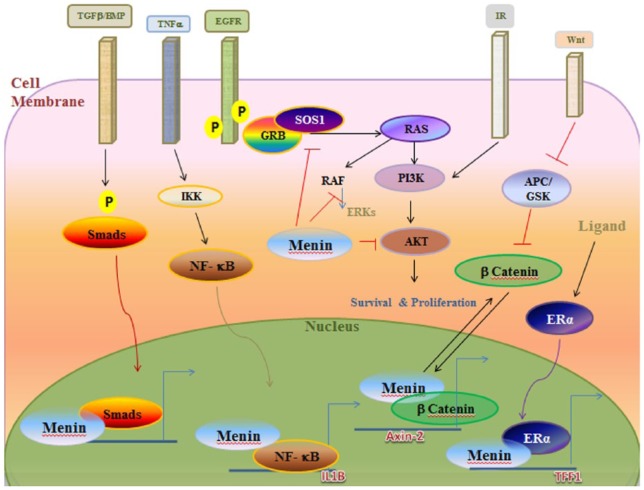
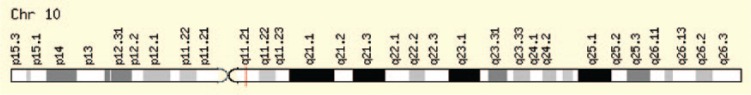
{"title":"Multiple Endocrine Neoplasia Syndromes from Genetic and Epigenetic Perspectives.","authors":"Fatemeh Khatami, Seyed Mohammad Tavangar","doi":"10.1177/1177271918785129","DOIUrl":null,"url":null,"abstract":"<p><p>Multiple endocrine neoplasia (MEN) syndromes are infrequent inherited disorders in which more than one endocrine glands develop noncancerous (benign) or cancerous (malignant) tumors or grow excessively without forming tumors. There are 3 famous and well-known forms of MEN syndromes (MEN 1, MEN 2A, and MEN 2B) and a newly documented one (MEN4). These syndromes are infrequent and occurred in all ages and both men and women. Usually, germ line mutations that can be resulted in neoplastic transformation of anterior pituitary, parathyroid glands, and pancreatic islets in addition to gastrointestinal tract can be an indicator for MEN1. The medullary thyroid cancer (MTC) in association with pheochromocytoma and/or multiple lesions of parathyroid glands with hyperparathyroidism can be pointer of MEN2 which can be subgrouped into the MEN 2A, MEN 2B, and familial MTC syndromes. There are no distinct biochemical markers that allow identification of familial versus nonfamilial forms of the tumors, but familial MTC usually happens at a younger age than sporadic MTC. The <i>MEN1</i> gene (menin protein) is in charge of MEN 1 disease, CDNK1B for MEN 4, and RET proto-oncogene for MEN 2. The focus over the molecular targets can bring some hope for both diagnosis and management of MEN syndromes. In the current review, we look at this disease and responsible genes and their cell signaling pathway involved.</p>","PeriodicalId":47060,"journal":{"name":"Biomarker Insights","volume":"13 ","pages":"1177271918785129"},"PeriodicalIF":2.6000,"publicationDate":"2018-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7b/c5/10.1177_1177271918785129.PMC6043927.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomarker Insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/1177271918785129","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Multiple endocrine neoplasia (MEN) syndromes are infrequent inherited disorders in which more than one endocrine glands develop noncancerous (benign) or cancerous (malignant) tumors or grow excessively without forming tumors. There are 3 famous and well-known forms of MEN syndromes (MEN 1, MEN 2A, and MEN 2B) and a newly documented one (MEN4). These syndromes are infrequent and occurred in all ages and both men and women. Usually, germ line mutations that can be resulted in neoplastic transformation of anterior pituitary, parathyroid glands, and pancreatic islets in addition to gastrointestinal tract can be an indicator for MEN1. The medullary thyroid cancer (MTC) in association with pheochromocytoma and/or multiple lesions of parathyroid glands with hyperparathyroidism can be pointer of MEN2 which can be subgrouped into the MEN 2A, MEN 2B, and familial MTC syndromes. There are no distinct biochemical markers that allow identification of familial versus nonfamilial forms of the tumors, but familial MTC usually happens at a younger age than sporadic MTC. The MEN1 gene (menin protein) is in charge of MEN 1 disease, CDNK1B for MEN 4, and RET proto-oncogene for MEN 2. The focus over the molecular targets can bring some hope for both diagnosis and management of MEN syndromes. In the current review, we look at this disease and responsible genes and their cell signaling pathway involved.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
